Journal of Chromatography & Separation Techniques
Open Access
ISSN: 2157-7064
ISSN: 2157-7064
Research Article - (2018) Volume 9, Issue 6
The paper reports an efficient and rapid fast LC method for estimation of diclofenac potassium and its impurities. The main aim of the study is to develop and validate a simple, rapid, accurate, sensitive, less time consuming and less expensive method by using RP-HPLC with UV detector. The chromatographic separation of diclofenac potassium and its impurities was carried out by using 150 × 4.6 mm, i.d., 5 μm C-18 column with prepared mobile phase-A consisting 800:200 (v/v) of 0.01 M ammonium acetate adjusted pH 5.3 with acetic acid and acetonitrile and mobile phase-B consisting 200:800 (v/v) of 0.01 M ammonium acetate adjusted pH 5.3 with acetic acid and acetonitrile. The wavelength for detection of were made at 280.0 nm using UV detector. The flow rate of 1 mL/min. The method was able to detect and separate impurities and diclofenac potassium. The system suitability parameters were found within the limits. The coefficient of correlation for diclofenac potassium and its impurities was found not less than 0.998. The %recovery was found within the ICH limits. The LOD and LOQ values from study demonstrate method is sensitive. The method was validated for linearity, precision, accuracy, specificity, intermediate precision, ruggedness, robustness, stability and suitability.
Keywords: Diclofenac potassium; Method development; Method validation; RP-HPLC method
Diclofenac (DIC) (2-[(2,6-dichlorophenyl)amino]phenyl acetic acid is an essential non-steroidal anti-inflammatory drug (NSAID) which is clinically prescribed for the treatment of inflammatory disorders, such as ankylosing spondylitis, osteoarthritis, and rheumatoid arthritis. On the other side, increased risk of serious cardiovascular thrombotic events, such as stroke and myocardial infarction are caused by excessive doses of this drug. Moreover, impurity profiling [1-3] of drug substances and drug products is critical tasks for pharmaceutical analytical scientist under regulatory industrial conditions. The presence of unwanted chemicals, by products and unknown impurities, even in very low levels, may change or effect the therapeutic efficacy and the safety of the pharmaceutical drug substances and drug products. Therefore, the impurity profile study of drug substances and drug products has to be carried out using a suitable validated analytical method in the final pharmaceutical industry.
High Performance Liquid Chromatography (HPLC) is one of the most effective analytical tools to determine quantitatively. Many individual components present in mixture via single analytical method. Also, these methods can separate a mixture of compounds to identify, quantify and purify the individual components of the mixture. Therefore, several research groups devoted their work to develop HPLC based methods for the determination and quantification of diclofenac [4-13]. In some literature diclofenac was determined in human plasma and other biological fluids with the help of different analytical techniques [14-19].
The current work demonstrates the development and validation of a rapid, simple, sensitive and efficient HPLC method for the determination of diclofenac potassium and its related impurities (Figure 1) with run time of 15 min. The analytical method separates all organic impurities and degradation products. Moreover, we successfully validated various parameters such as system suitability, specificity, precision, accuracy, linearity, ruggedness and robustness. The limit of detection and limit of quantification of this method was found to be 0.083 ppm and 0.253 ppm respectively.

Figure 1: Structures (A) Diclofenac Potassium, (B) Oxindole, (C) Impurity-A, (D) Impurity-D, (E) Alcohol analog, (F) Benzaldehyde analog.
Instruments, chemical and reagents
The waters HPLC (Model: 2695) with PDA, Metler Toledo analytical balance, ultra-sonic bath, centrifuge (model Beckman J2) were used for current analysis. Analytical reagents were of analytical HPLC grade used. High purity water was taken from Evoqua water purification system. Analytical grade ammonium acetate, acetic acid were purchased from the S.D. Fine Chem. Pvt. Ltd., HPLC grade acetonitrile was procured from RANKEM India Pvt limited, India. Working standard was obtained from Aurex Laboratories Pvt Ltd, Hyderabad and impurities are purchased form Simson Pharma Mumbai India.
Analytical conditions
The waters HPLC (Model: 2695) with PDA used consists of a Quaternary solvent manager, a sample manger and a photodiode array UV detector. The output signal was monitored and processed using Empower 3 software. The HPLC separation was carried out on a Zodiac 150 × 4.6 mm, i.d., 5 μm column was used as a stationary maintained at 35°C. The mobile phase was Prepared buffer with ammonium acetate (0.1 M) and adjusted pH 5.3 with Acetic acid. Prepared mobile phase-A with buffer: acetonitrile in the ratio of 800:200 and Mobile phase-B with buffer and acetonitrile in the ratio 200:800 then filtered through 0.45 μm PTFE filter and degassed in an ultra sonicator for about 15 min prior to use, the flow rate of 1 mL min-1. The wavelength for detection of compounds was made at 280.0 nm using PDA, injection volume was 10.0 μL.
Preparation of analytical solutions
Preparation of buffer: Prepared (0.1 M) ammonium acetate in 1000 mL of purified water, adjusted pH 5.3 with acetic acid and filtered through the 0.45 μm filter.
Preparation of diluent: A mixture of water and acetonitrile (50:50, v/v %), was used as the diluents for throughout the experiments. All the solutions and solvents sonicated and filtered through 0.45 μm membrane filters prior to the analysis.
Preparation of standard and sample: To prepare the sample taken 20 tablets weighed on the analytical balance and calculated the average weight and transferred to mortar vessel crushed in to a fine powder and mix well to get the uniformity of the powder. After the sample and standard stock solutions are prepared by dissolving in 20% of diluent and made up to volume with diluent. The concentrations of diclofenac potassium are 500 μg mL-1 and 50 μg mL-1 for related substances and Assay methods, respectively. Diluted 500 μg mL-1 related substances sample to get 50 μg mL-1 for assay determinations. Prepare known concentrations of impurity stock solutions and then spike the sample to perform precision, accuracy, LOD, LOQ and linearity parameters.
Chromatographic conditions for HPLC
A Zodiac C18 (150 mm × 4.6 mm, i.d., 5 μm), mobile phase-A as a 0.1 M ammonium acetate as buffer and acetonitrile (80:20, v/v%), mobile phase-B, 0.1 M ammonium acetate as buffer and acetonitrile (20:80, v/v%). Flow rate of 1.0 mL min-1 with gradient elution at column temperature of 35°C. The Detection of the column effluents by PDA at 280 nm. The sample injection volume was 10 μL. The Gradient program is mentioned in Table 1.
| Time in min | Flow rate | % Mobile phase-A | % Mobile phase-B |
|---|---|---|---|
| 0.00 | 1.0 | 65 | 35 |
| 7.00 | 1.0 | 65 | 35 |
| 9.00 | 1.0 | 0 | 100 |
| 13.00 | 1.0 | 0 | 100 |
| 13.10 | 1.0 | 65 | 35 |
| 15.00 | 1.0 | 65 | 35 |
Table 1: Gradient method.
Specificity and stress degradation studies
Diclofenac Potassium RS sample solution (Concentration 500 μg mL-1) was used to perform all stress degradation studies [20]. In each condition the diclofenac potassium API sample 50 mg transferred in to 100 mL volumetric flask, acid and alkaline hydrolysis were executed by heating the drug solutions in 5 mL of HCl and 1 N NaOH respectively at 60°C for 1 hour. Stress testing for neutral condition was carried out by heating the drug dissolved in water at 60°C for 1 hour. For doing study in oxidative condition, the drug solution was kept at 60°C in 5 mL of 30% hydrogen peroxide for 2 hours. For photolytic study, the drug sample was exposed to UV Light (200-Watt hours/m2), for visible light 1.2 million Lux hours in the photo stability chamber for about 24 hours. Additionally, the drug solution was exposed to dry heat at 60°C for 24 hours in a hot air oven to perform the thermal degradation study. After the stipulated time, the samples were withdrawn and subjected to HPLC analysis soon after suitable dilution was made with diluent (500 μg mL-1) and for acid and base stress samples are neutralized after that made suitable dilutions. To calculate the mass balance for each condition of the RS sample (Concentration 500 μg mL-1) was diluted to assay concentration (50 μg mL-1) and subjected to HPLC analysis.
Method development
The HPLC method was optimized with a view to develop a stability indicating assay method, which is capable of eluting and resolving diclofenac potassium from its impurities. USP official method is available in the with UPLC method with 15 minutes run time. But the organization will not able to purchase the UPLC system because of the cost. More over the columns which are used in the UPLC method also very expensive. By using the waters software same programme was converted in to HPLC method. Conversion of UPLC method to HPLC method by using waters software (Table 2).
| Parameters | UPLC method | HPLC method | ||||
|---|---|---|---|---|---|---|
| Detector Wavelength | 280 nm | 280 nm | ||||
| Flow rate in mL/min | 0.30 | 0.52 | ||||
| Column dimensions | C18,100 mm × 2.0 mm, 1.9 µm | C18,150 mm × 4.6 mm, 5.0µm | ||||
| Column Temperature | 35°C | 35°C | ||||
| Injection volume | 1 µL | 7.2 µL | ||||
| Gradient Programme | Time | MP-A | MP-B | Time | MP-A | MP-B |
| 0 | 70 | 30 | 0 | 70 | 30 | |
| 0.5 | 70 | 30 | 1.89 | 70 | 30 | |
| 8.5 | 5 | 95 | 35.72 | 5 | 95 | |
| 10.0 | 5 | 95 | 41.47 | 5 | 95 | |
| 10.1 | 70 | 30 | 41.51 | 70 | 30 | |
| 14 | 70 | 30 | 58.14 | 70 | 30 | |
Table 2: Conversion of UPLC method to HPLC method by using waters software.
Analytical method validation
The validation of the HPLC method was carried out for the estimation of related substances (i.e., Impurity-A, Oxinidole, alcohol analogue, Impurity-D and benzaldehyde analogue) and assay of diclofenac potassium as per the ICH guidelines [21] to demonstrate that the method is appropriate for intended use.
System suitability: The system suitability test was performed before starting every validation parameter by injecting the 5-μg mL-1 and 50 μg mL-1 of diclofenac potassium solution for the estimation of related substance and assay methods, respectively.
Specificity: The specificity of the method was determined by the preparation of blank, placebo solution and individual impurities of diclofenac potassium were prepared with the known concentration and subjected to the HPLC analysis.
Linearity: Linearity of the developed method for the impurities was established by analyzing a series of five known concentration levels ranging from 1 μg mL-1 to 15 μg mL-1 of impurities spiked into the diluent for RS method, the calibration curves were prepared by plotting the peak areas of the impurities against their corresponding concentrations. Similarly, assay method linearity was established by injecting diclofenac potassium at five different concentration levels of 12 μg mL-1 to 80 μg mL-1. The correlation coefficients (r2), slopes and Y-intercepts of diclofenac potassium and its impurities were calculated from their respective calibration plots.
Precision: The HPLC system precision were verified by analyzing the six replicates of standard solution of both assay (Diclofenac potassium 50 μg mL-1) and related substances (Diclofenac potassium 5 μg mL-1) method individually. The method precision for related substances were evaluated by injecting the six individual test preparations of diclofenac potassium for assay (50 μg mL-1) and diclofenac potassium (500 μg mL-1) spiked with 0.2% each impurity, respectively. The intermediate precision was evaluated at the same concentrations prepared separately on different days by different analysts. Precision at LOQ levels was also determined by injecting six individual preparations of mixtures of all impurities spiked into diclofenac potassium sample at their LOQ level. The %RSDs of peak areas of diclofenac potassium and its impurities were calculated for precision studies.
Accuracy: Method accuracy expressed in terms of recovery percentage was assessed by analyzing samples with known concentrations of diclofenac potassium and its impurities and calculating the percentage of the recovered amounts. The recovery tests were carried out in triplicates at three concentration levels (50%, 100% and 150%), with impurities concentration of 0.5, 1.0, and 1.5 μg mL-1 each (i.e., 0.2%with respect to Diclofenac potassium concentration (500 μg mL-1). The accuracy of the diclofenac potassium assay was evaluated in triplicate at three concentration levels of 25, 50 and 75 μg mL-1, and the recovery was calculated for each concentration.
Limits of Detection (LOD) and Quantitation (LOQ): The LOD and LOQ values for diclofenac potassium and its impurities (i.e., Impurity-A, Oxinidole, alcohol analogue, Impurity-D and Benzaldehyde analogue) were determined based on the standard deviation of response and slope found respectively.
Robustness: Robustness of the developed LC method was established by evaluating the effect of small variations in the optimized chromatographic conditions such as flow rate (± 0.2 mL.min-1), column temperature (± 5°C), pH of buffer (± 0.2) and mobile phase composition (± 10.0%) while keeping the components of the mobile phase constant. Performed filter compatibility with Nylon and PVDF for spiked sample and compared with centrifuged sample.
Forced degradation studies were conducted for diclofenac potassium solid dosage formulation and placebo to identify the specificity of the assay and RS method. The FDS conducted on the samples using the ICH Q1 A (r2) guidelines acidic, alkaline, oxidative, photolytic, and thermal degradation. The diclofenac and its impurities peak purity was passed. The degradation peaks were effectively separated from the main peak and its known impurities in this method. The forced degradation conditions are shown in Table 3.
| Stress Condition | Solvent | Temperature (°C) | Sampling time (h) |
|---|---|---|---|
| Hydrolytic (Neutral) | H2O | 60 | 1 |
| Acid | 1 N HCl | 60 | 1 |
| Basic | 1N NaOH | 60 | 1 |
| Oxidizing | 30% H2O2 | 60 | 2 |
| Thermal | Diluent* | 60 | 24 |
| Photolytic condition | Diluent* | - | 24 |
*Acetonitrile and water (50:50 v/v), h=hours
Table 3: Stress conditions for Diclofenac Potassium.
Acid, alkali and water forced degradation (Hydrolytic conditions): The sample (Concentration 500 μgmL-1) was transmitted to each of three 100 mL volumetric flasks and the volume 5 mL of 1.0 N HCl, 1.0 N NaOH and water was added and then kept on Heating water bath at 60°C for about 1 hour. After the stipulated time, the samples were withdrawn and neutralized to get the neutral pH 7.0 then subjected to HPLC analysis soon after suitable dilution individually data are shown in Table 3.
Hydrogen peroxide-forced degradation (Oxidizing conditions): The Sample (Concentration 500 μgmL-1) was transferred to 100 mL volumetric flasks and the volume 5 mL of 10% H2O2, added then kept on Heating water bath at 60°C for about 2 hours. The samples were withdrawn and subjected to HPLC analysis soon after suitable dilution data are shown in Table 3.
Thermal degradation: The Sample (Concentration 500 μgmL-1) transferred to each of 100 mL volumetric flasks and the volume was made up to the mark with diluents which were exposed to heat 60°C for 24 h in a convection oven respectively, data are presented in Table 3.
Photolytic degradation: The Sample (Concentration 500 μgmL-1) was transferred to a 100 mL volumetric flask were exposed UV light and Visible light for 24 hours in the photo stability chamber then the volume was made up to the mark with diluents, data are presented in Table 3.
HPLC method development
In order to optimize the method, different experimental conditions were evaluated such as buffer concentration, organic solvent and other chromatographic conditions using C18 column.
Firstly, we optimized the type and concentration of buffer solution. Ammonium acetate was selected as buffer solution because of its volatile nature. 0.1 M, 0.2 M and 0.3 M concentrations of ammonium acetate was used to investigate the suitable buffer concentration. During the experiment we observed that, the chromatograms are similar with these three buffer concentrations. Therefore, we used lowest concentration of ammonium acetate i.e., 0.1 M. Next, we used methanol and acetonitrile solvents to optimize efficient organic solvent to obtained symmetry and better peak for the analytes. The experimental results showed that, both solvents give similar symmetry and peak shape. The experimental results indicated, in case of acetonitrile solvents give faster elution peaks. The gradient program consisting of 0.0-7.0 min 35% mobile phase B and 7.0-9.0 min gradient up to 100% mobile phase B, this was continued up to 13 min. After this gradient was returned to the initial stage.
Method validation
Various parameters were validated such as system suitability, specificity, linearity, Limit of Detection (LOD) and Limit of Quantification (LOQ), accuracy, precision, and robustness to perform and prove that the development method is capable to generate accurate and reproducible results.
System suitability: The system suitability data indicating that the HPLC system was suitable for method validation the tailing factor for all the analytes was less than 1.5 and the resolution between any of the two adjacently eluting analytes were greater than 2. The %relative standard deviation of six injection of assay standard found below 2.0% and RS standard below 5.0% It also confirms that the good selectivity of the method, data are presented in Table 4. Chromatograms are shown in Figure 2.

Figure 2: Reverse phase HPLC chromatograms obtained for the Blank (A), Chromatogram of Diclofenac Potassium (Conc. 50 μg mL-1). (C) Chromatogram of formulation product.
| Parameter (n=5) | Results (Mean ± SD, RSD) |
Required Limits |
|---|---|---|
| Retention time in minutes (Rt) | 9.576 ± 0.01, 0.16 | RSD ≤ 2 |
| Theoretical plates (N) | 8424.8 ± 58, 0.7 | N>2500 |
| Tailing Factor (T) | 1.15 ± 0.01, 1.23 | T≤ 2 |
Table 4: System Suitability Results.
Specificity: Specificity was established by injecting a mixture of diclofenac potassium (500 μgmL-1) with impurities 1.0 μgmL-1 (0.2% of each impurity). Placebo and diluent solution have no interference at the retention time of diclofenac potassium and its impurities, which proved that the method is sufficiently specific.
Linearity: Linearity results of diclofenac potassium and related substances showed the existence of an excellent correlation (r2>0.999) between the peak area and the concentrations. The Calibration curves are shown in Figure 3A-3G.

Figure 3: Calibration curve of Oxindole (A), Impurity-A (B), Alcohol analog (C), Impurity-D (D), Benzaldehyde analog (E), Diclofenac Potassium RS (F), Diclofenac Potassium AY (G).
Precision: The %Relative standard deviation values for both precision values were less than 2.0% for assay method, The %relative standard deviation values for both precision values are less than 5.0% for RS method, indicating good precision of the method, chromatograms are shown in Figure 4 and Table 5.

Figure 4: Reverse phase HPLC chromatogram Spiked impurities sample.
| S. No. | % of impurities | ||||||
|---|---|---|---|---|---|---|---|
| Oxindole (Imp-E) | Imp-A | Alcohol analog | Imp-D | Benzaldehyde Analog | Diclofenac Potassium (RS) |
Diclofenac Potassium (AY) |
|
| Sample 1 | 0.210 | 0.214 | 0.201 | 0.198 | 0.201 | 0.205 | 98.1 |
| Sample 2 | 0.208 | 0.210 | 0.202 | 0.201 | 0.201 | 0.201 | 98.0 |
| Sample 3 | 0.201 | 0.208 | 0.201 | 0.205 | 0.205 | 0.202 | 98.1 |
| Sample 4 | 0.205 | 0.205 | 0.205 | 0.201 | 0.201 | 0.200 | 98.3 |
| Sample 5 | 0.205 | 0.208 | 0.201 | 0.195 | 0.206 | 0.208 | 98.1 |
| Sample 6 | 0.208 | 0.206 | 0.201 | 0.198 | 0.208 | 0.206 | 98.2 |
| Average | 0.206 | 0.209 | 0.202 | 0.200 | 0.204 | 0.20 | 98.1 |
| %RSD | 1.55 | 1.54 | 0.79 | 1.73 | 1.51 | 1.54 | 0.11 |
| Intermediate Precision | |||||||
| Sr.No. | Oxindole (Imp-E) | Imp-A | Alcohol analogue | Imp-D | Benzaldehyde Analogue | Diclofenac Potassium (RS) |
Diclofenac Potassium (AY) |
| Sample 1 | 0.201 | 0.208 | 0.203 | 0.205 | 0.199 | 0.201 | 101.2 |
| Sample 2 | 0.205 | 0.201 | 0.201 | 0.201 | 0.198 | 0.203 | 102.6 |
| Sample 3 | 0.206 | 0.203 | 0.202 | 0.205 | 0.198 | 0.201 | 100.6 |
| Sample 4 | 0.201 | 0.201 | 0.201 | 0.205 | 0.205 | 0.206 | 101.0 |
| Sample 5 | 0.202 | 0.206 | 0.206 | 0.206 | 0.201 | 0.201 | 102.6 |
| Sample 6 | 0.206 | 0.201 | 0.201 | 0.198 | 0.200 | 0.201 | 99.4 |
| Average | 0.204 | 0.203 | 0.202 | 0.203 | 0.200 | 0.20 | 101.2 |
| %RSD | 1.19 | 1.48 | 0.97 | 1.53 | 1.32 | 1.01 | 1.2 |
Table 5: Precision studies for Diclofenac Potassium and its impurities.
Limit of detection and limit of quantification: The Limit of Detection (LOD) and Limit of Quantification (LOQ) of diclofenac and its impurities were established by slope method. In the slope method, prepared series of solution from low concentration to high concentration of diclofenac and impurities in the diluent and subjected to the HPLC. LOD and LOQ were calculated by using the following formula.
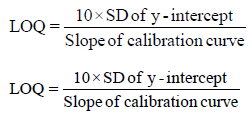
LOD and LOQ results of diclofenac and its related substances, indicating the higher sensitivity of the method. The results of LOD, LOQ, precision, and linearity are summarized in Tables 6A and 6B.
| S. No | Impurity Name | Slope | Intercept | SD (STEX) | LOD ppm |
LOQ ppm |
RRF |
|---|---|---|---|---|---|---|---|
| 1 | Oxinidole | 35816.09 | 1223.5 | 1767.115 | 0.163 | 0.493 | 4.49 |
| 2 | Impurity-A | 15054.89 | 89.49 | 451.653 | 0.099 | 0.300 | 1.89 |
| 3 | Alcohol Analog | 9024.57 | 57.6 | 591.408 | 0.216 | 0.655 | 1.13 |
| 4 | Diclofenac | 7985.73 | 138.83 | 202.061 | 0.083 | 0.253 | 1.00 |
| 5 | Impurity-D | 4456.255 | 6.885 | 113.825 | 0.084 | 0.255 | 0.56 |
| 6 | Benzaldehyde analog | 18982.55 | 989.55 | 466.664 | 0.081 | 0.246 | 2.38 |
Table 6A: LOD and LOQ of Establishment for Diclofenac Potassium and its impurities.
| S. No. | % of Impurities at LOQ level | |||||
|---|---|---|---|---|---|---|
| Oxindole (Imp-E) | Imp-A | Alcohol analog | Imp-D | Benzaldehyde Analog | Diclofenac Potassium | |
| Sample 1 | 0.099 | 0.061 | 0.125 | 0.051 | 0.048 | 0.042 |
| Sample 2 | 0.095 | 0.065 | 0.120 | 0.049 | 0.048 | 0.044 |
| Sample 3 | 0.102 | 0.065 | 0.118 | 0.050 | 0.049 | 0.045 |
| Sample 4 | 0.101 | 0.061 | 0.128 | 0.055 | 0.051 | 0.044 |
| Sample 5 | 0.099 | 0.062 | 0.121 | 0.042 | 0.051 | 0.041 |
| Sample 6 | 0.098 | 0.063 | 0.125 | 0.048 | 0.048 | 0.042 |
| Average | 0.099 | 0.063 | 0.123 | 0.049 | 0.049 | 0.04 |
| %RSD | 2.47 | 2.92 | 3.06 | 8.67 | 2.99 | 3.60 |
Table 6B: LOQ Precision for Diclofenac and Its impurities.
Accuracy: The mean percentage recoveries of impurities and diclofenac potassium determining the developed method is suitable to estimate the diclofenac potassium and its impurities. data are presented in Table 7.
| Imp. Name | Level (n=3) |
‘µg/mL’ added |
‘µg/mL’ found |
% Recovery (n=3) |
% RSD (n=3) |
|---|---|---|---|---|---|
| Oxindole | 50% | 0.4990 | 0.5083 | 101.9 | 1.4 |
| 100% | 0.9980 | 0.9917 | 99.4 | 1.5 | |
| 150% | 1.4970 | 1.4750 | 98.6 | 1.5 | |
| Impurity-A | 50% | 0.5010 | 0.5042 | 100.6 | 2.9 |
| 100% | 1.0020 | 0.975 | 98.6 | 2.2 | |
| 150% | 1.5030 | 1.4875 | 99.0 | 1.5 | |
| Alcohol Analog | 50% | 0.5010 | 0.5083 | 101.5 | 1.4 |
| 100% | 1.0020 | 0.9917 | 99.0 | 0.7 | |
| 150% | 1.5030 | 1.4917 | 99.3 | 0.5 | |
| Impurity-D | 50% | 0.5010 | 0.5125 | 102.3 | 2.4 |
| 100% | 0.9980 | 1.0001 | 100.2 | 1.3 | |
| 150% | 1.5040 | 1.4875 | 98.9 | 0.8 | |
| Benzaldehyde analog | 50% | 0.5040 | 0.5167 | 102.5 | 1.4 |
| 100% | 1.0070 | 1.0125 | 100.5 | 2.4 | |
| 150% | 1.5110 | 1.5208 | 100.6 | 3.3 | |
| Diclofenac (RS) | 50% | 0.5030 | 0.5000 | 99.4 | 2.5 |
| 100% | 1.0060 | 0.9833 | 97.8 | 0.50 | |
| 150% | 1.5090 | 1.4792 | 98.0 | 0.50 | |
| Diclofenac (AY) | 50% | 25.1510 | 24.8625 | 98.9 | 1.00 |
| 100% | 50.2990 | 50.5917 | 100.6 | 1.00 | |
| 150% | 75.4490 | 76.3542 | 101.2 | 0.90 |
Table 7: Accuracy studies for Diclofenac Potassium and its impurities.
Robustness: In all deliberately altered chromatographic conditions (flow rate, pH and column temperature), all analytes were adequately resolved, and elution order remains unchanged. The resolutions between any two adjacent analytes were obtained greater than 1.5 and tailing factors of all analytes were obtained less than 1.8. Filter compatibility Nylon and PVDF results are comparable with centrifuged sample indicating the robustness of the method.
Stabilities
Stability of the drug product was conducted at different conditions for Quality Control (QC) of samples. The assay and RS samples were compared with freshly analyzed QC samples, in that we found no difference was found in % impurities and % of Assay. So that no degradation of product while doing the analysis. The Standard and sample were stable at room temperature for about 48 hours.
Forced degradation studies
The objective of the forced degradation studies was to determine the intrinsic stability of the drug product and specificity of the method under different conditions as 1 N HCl, 1 N NaOH, 30% H2O2, exposed to heat (thermal energy) or radiation (photo light) and neutral. Each condition the chromatograms are recorded and peak area, % of the drug degraded was calculated. The present investigation, it was concluded that drug was stable in Base, thermal, and photolytic and unstable in acid, and oxidation. The percentage of the degradations was 10% in the oxidation. The experimental results were given in Table 8 and chromatograms were presented from Figure 5A-5G.

Figure 5: Reverse phase HPLC chromatogram and Purity plot of Control sample (A), Acid Stressed sample (B), Base Stressed sample (C), Peroxide Stressed (D), Heat Stressed (E), Water Stressed (F), Photo stability Stressed (G).
| S. No. | Stress Condition | Assay | % of Impurities | Mass Balance | Purity Angle | Purity Threshold | Purity Flag |
|---|---|---|---|---|---|---|---|
| 1 | Controlled sample | 99.82 | 0 | NA | 0.213 | 0.380 | NO |
| 2 | Acid Hydrolysis | 94.2 | 3.9 | 98.1 | 0.487 | 1.318 | NO |
| 3 | Base Hydrolysis | 98.1 | 1.3 | 99.4 | 0.127 | 0.292 | NO |
| 4 | Oxidation | 69.8 | 26.52 | 96.3 | 0.132 | 0.324 | NO |
| 5 | Thermal | 99.2 | 0 | 99.2 | 0.257 | 0.367 | NO |
| 6 | Neutral | 99.9 | 0 | 99.9 | 0.252 | 0383 | NO |
| 7 | Photolytic | 99.7 | 0 | 99.7 | 0.213 | 0.380 | NO |
Table 8: Forced degradation Results for diclofenac potassium.
The major challenges were faced in the current study, First, the resolution between the impurities with less run time. Sensitivity of impurities at low concentration. Selected appropriate mobile phase composition and gradient method to get the optimum resolution between the impurity peaks. Optimized the test concentration to get the desired LOQ levels for the impurities and diclofenac. Therefore, the aim of the method development completed successfully.
According the USP 621 General chapter system suitability is basic criteria for the liquid chromatography. The current optimized method system suitability results found satisfactory.
Method validation was performed as per the ICH guidelines [21]. The linearity of the proposed HPLC method was performed for the diclofenac and its impurities. Linear relationships between Diclofenac and its impurities and concentration and high regression coefficients were obtained for each impurity. Accuracy of the proposed method was assessed by calculating %recovery of four different concentrations (LOQ, 50%, 100% and 150%) of laboratory prepared mixtures of the impurities and diclofenac analyzed by the proposed method. The accuracy was also confirmed by recovery studies from sample solution at different levels of standard additions. Intra-day and inter-day precision were also evaluated. In addition, good results in terms of LOD, LOQ, robustness and selectivity were obtained.
The Current paper is an accurate, specific and reproducible stability-indicating RP-HPLC Assay and RS method was developed and validated for the quantification of diclofenac and its impurities according to ICH and USP guidelines. The HPLC method shows good performance with linearity, accuracy, precision, specificity and robustness. The Method have high sensitivity for the impurities and high percentage recoveries were obtained by this method. This is the first stability indicating method developed and have the capability to resolve all the diclofenac degradation products from the drug substance.
The authors wish to thank the management of Aurex Laboratories and GITAM for supporting this work and guidance is appreciated.