Journal of Chromatography & Separation Techniques
Open Access
ISSN: 2157-7064
ISSN: 2157-7064
Research Article - (2014) Volume 5, Issue 2
An analytical method combining off-line solid phase extraction (SPE) and reversed phase high performance liquid chromatography (HPLC) has been developed for simultaneous determination of six herbicides. The compounds analyzed in natural surface water samples, were: the herbicides atrazine (AT), simazine (SI) and their major dealkylated and hydroxylated degradation products such as deisopropyl-atrazine (DIA), deethyl-atrazine (DEA), hydroxyl-atrazine (H-AT) and hydroxyl-simazine (H-SI), all of them with herbicide activity too. As optimum conditions, a Kromasil C18 column as stationary phase, a mixture of acetonitrile and phosphate buffer pH 7.0 as mobile phase in gradient elution mode and 215 nm as measuring wavelength, were selected. A polymeric cartridge (Bond-Elut ENV-1000 mg) was used for a SPE procedure. The optimized off-line SPE HPLC-DAD method was evaluated in terms of validation and robustness procedure, obtaining among other parameters, limits of quantification between 81 and 100 ng L-1, considering the enrichment factor for all of the studied herbicides. The proposed method was successfully applied to the screening of herbicides in natural surface water samples from different locations in the 23th aquifer (Albacete, Spain).
Keywords: S-Triazines; Degradation products; Solid-phase extraction; High performance liquid chromatography; Natural surface waters from aquifer
Pesticides such as s-triazines are widely used as selective pre and post-emergence herbicides for the control of broadleaved, grassy areas, in many agricultural crops like corn, wheat, as well as in railways, roadside verges and golf courses. After application of triazines, several degradation processes take place leading to dealkylation of amine groups (positions 4 and 6 of the triazinic structure), or hydrolysation of the substituent in position 2 according to the chemical structures shown in Table 1.
| Pesticides | Chemical Structures | pKa |
|---|---|---|
| Atrazine (AT) | 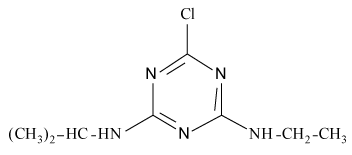 |
1.68-1.85 |
| Simazine (SI) | 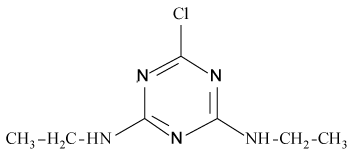 |
1.65-1.80 |
| Deethyl-atrazine (DEA) | 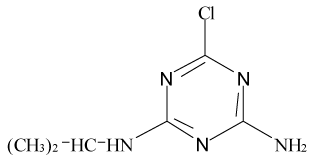 |
1.65 |
| Deisopropyl-atrazine (DIA) | 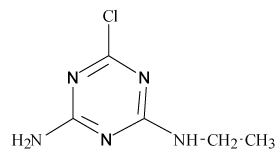 |
1.58 |
| Hydroxyl-atrazine (H-AT) | 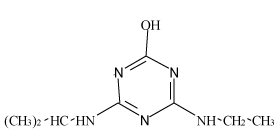 |
5.2 |
| Hydroxyl-simazine (H-SI) | 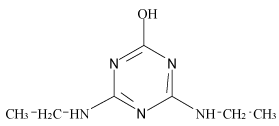 |
5.2 |
Table 1: Chemical structures and pKa values of studies analytes.
Atrazine (AT), (2chloro-N4-ethyl-N6-isopropyl-1,3,5-triazine- 4,6-diamine), Simazine (SI), (2chloro-N4,N6-diethyl-1,3,5-triazine- 4,6-diamine) and their main dealkylated and hydroxylated degradation products, Deethyl-atrazine (DEA), (dealkylated degradation product obtained by losing of ethyl group in N4 amine function of Atrazine structure) Deisopropyl-atrazine (DIA), (dealkylated degradation product obtained by losing of isopropyl group in N6 amine function of Atrazine structure) hydroxyl-atrazine (H-AT) (degradation product obtained by replacing the Cl substituent at position 2 by hydroxyl group in Atrazine structure) and hydroxyl-simazine (HSI), (degradation product obtained by replacing the Cl substituent at position 2 by hydroxyl group in Simazine structure) are considered inside to the group of endocrine-disrupting chemicals [1], according to U.S. Environmental Protection Agency (EPA). The EPA defines an environmental endocrine or hormone disrupting chemical as an exogenous agent, which interferes with the synthesis, secretion, transport, binding, action or elimination of natural hormones in the body, and that are responsible for the maintenance of homeostasis, reproduction, development and/or behavior [2].
Both atrazine, simazine and their degradation products, are relatively persistent making necessary their analysis and quantification in environmental samples.
The continuous monitoring of pesticides and degradation products in natural waters, is a great importance matter to control the toxicity of the environment. Nowadays, water quality receives considerable attention for its obvious implication in public health, so, stringent regulations have been issued by legislation agencies. In this sense, current European Union (EU) directives [3], dictates that concentrations of individual pesticides must not exceed maximal admissible values of 0.1 μg L-1 in drinking water, whereas in natural surface waters, the alert and alarm threshold values are typically 1 and 3 μg L-1, respectively.
In the literature, most of the reported methods for quality determination and quantification of s-triazines in water samples, involves separation by high performance liquid chromatography (HPLC) or gas chromatography (GC) after liquid-liquid (LLE) [4,5], solid phase extraction (SPE) [6,7], or either more currently using liquid-liquid microextraction or solid-phase microextraction [8,9]; being well-known the advantages of SPE technique over LLE. Therefore, SPE procedures using C18-silica, polymeric sorbents, (i.e. nonpolar polystyrene-divinylbenzene), or other similar matrixes [10], for the analysis of both organic contaminants in water samples [11], and determination of triazines in environmental samples [12-14], have been previously reported. Other kinds of materials as multiwalled carbon nanotubes (MWCN) have also been successfully used with exceptional merit as SPE sorbents for enrichment of environmental pollutants [15]. Currently, Al-Degs et al. [16], developed a sensitive multiresidue procedure for the analysis of four common pesticides, where two of them were SI and AT, using off-line SPE-HPLC-UV and MWCN as adsorbent for the preconcentration of these herbicides, in water samples.
However, few papers have been published related to the screening of the main degradation products, which most of them have got herbicide activity too [17,18].
The parent s-triazines SI and AT and with some restriction the dealkylated degradation products too, have been analyzed by GC using a mass spectrometry detector (MSD), or a nitrogen-phosphorus detector (NPD) [19-22]. In this way, Berzas et al. [23], separated and determinated nine common herbicides as SI, AT and their dealkylated degradation products DEA, DIA among other related herbicides compounds, using a very sensitive capillary gas chromatographic mass spectrometric-selective ion monitored (GC-MS-SIM) as analytical technique. Although it is well-known, GC-MS still remains as the most powerful technique to separate and quantify the above-cited herbicides, but if among the analytes are included their hydroxyl degradation products, they cannot be easily analyzed directly by GC, because of their high polarity and non-volatilily. Then, HPLC and capillary electrophoresis (CE) are other suitable alternative techniques, for the analysis of both s-triazines and their hydroxylated degradation products. In fact, a recent work by Carabias et al. [24] makes a comparison between non-aqueous capillary electrophoresis and HPLC methods, for the determination of several chloro-s-triazines, thiomethyl-s-triazines and three main degradation products of parent compound AT. This research concluded that both methods afford similar results for the analysis of surface and drinking water samples.
HPLC techniques present as great advantage to be directly applicable to both s-triazines and their OH degradation products, reporting in the literature couplings with different detection modes like UV [25], MS [26-28] or even MS-MS tandem [7,29]. On a similar way, a currently paper developed by Dos Santos et al. [30], describes a sequential injection chromatographic procedure for separation and quantification of the herbicides SI, AT and propazine, exploring also the low backpressure of a 2.5 cm long monolithic C18 column. Most of these methods are mainly focused on parent triazines, even in their dealkylated residues [28], but not in the hidroxylated ones until now.
Therefore, the development of analytical methods for screening the presence and amounts of herbicides and their residues in natural waters, is an obvious interesting matter to prevent toxicological risks.
In the present work, an easy and fast off-line SPE HPLC-DAD procedure has been proposed for the isolation, separation and quantification of parents herbicides SI, AT and their main dealkylated (DEA, DIA) and hydroxylated (H-AT, H-SI) degradation products, commonly found in natural water samples.
An extensive validation study, which includes an exhaustive design robustness procedure as novelty, has been also carried out to assess the reliability of the obtained results. Finally, the whole method was successfully applied to the screening of SI, AT and their main dealkylated and hydroxylated degradation products, in natural surface water samples from different soundings at 23rd aquifer (Albacete, Spain).
This final analysis using natural water samples proved the usefulness of our proposed method, using a simple technique with optical detection to analyze the targeted compounds at relevant levels. This is especially significant for hydroxylated degradation products, very difficult to analyze by other methods without a previous derivatization, but nevertheless, very common in these surface natural waters.
As another value of the method, the LOQs reached for studied herbicides (between 81 and 100 ng L-1), are from ten to thirty fold lower than the maximal allowed amounts for natural surface waters(1 and 3 μg L-1 as alert and alarm levels), according to European Union directives [3].
The added value of our method remains in its proved reliability, in opposite to others types of sorbents because of its solubility problems (MWCN), or the use of backpressure monolithic columns due to the difficult separated elution, which involves separation, quantification and detection problems. Besides, these recent approaches have been just used until now for the analysis of the parent compounds, but not for their degradation products yet.
Chemicals
All solvents and reagents were of analytical grade unless indicated otherwise. Acetonitrile (HPLC-grade) was obtained from Scharlau S.L. (Barcelona, Spain) and methanol (HPLC-grade) from Panreac (Barcelona, Spain). Buffer solutions were prepared using the following reagents: sodium dihydrogenphosphate, disodium hydrogenphosphate and phosphoric acid (HPLC-grade). All of these compounds were obtained from Merck (Darmstadt, Germany).
Simazine (SI) and atrazine (AT) were obtained from Riedel-de Haën (Seelze, Germany), whereas hydroxyl-atrazine (H-AT) and hydroxyl-simazine (H-SI), deisopropyl-atrazine (DIA) and deethylatrazine (DEA), were purchased from Dr. Ehrenstorfer (Augsburg, Germany).
Apparatus
The HPLC system utilized was a Shimadzu (Kyoto, Japan) model LC-10AD with a solvent delivery pump, a Rheodyne injection valve (20 μL sample loop) and a SPD-M10A photodiode-array detector. The system was controlled by a computer equipped with the CLASS-LC 10 software, which was used for all of the measurements and treatment of data.
Separation was achieved at room temperature using a reversedphase Kromasil C18 column (4.6 i.d. ×150 mm; particle size 5 μm), purchased from Waters (Milford, MA, USA). pH values were measured by using a Crison model 2001 pHmeter provided with a combined glass electrode.
Phosphate buffer solutions and water samples were filtered through a 0.45 μm MFTM-membrane filters, and acetonitrile was filtered through a 0.5 μm Fluoropore TM membrane filters. Both membrane filters were purchased from Millipore (Milford, MA, USA).
The extraction and pre-concentration processes were carried out using a device developed in our laboratories consisting of a water manifold, Visiprep TM Sep-Pack system, from Supelco (Madrid, Spain), coupled to a Vacuum pump XF 54 23050, from Millipore (Milford, MA, USA). The SPE procedure was performed using Bond Elut-ENV SPE Sep-Pack Plus cartridges, with styrene divinylbenzene co-polymer sorbent (1000 mg), from Water (Milford, MA, USA). Methanolic extracts were filtered with 0.45 μm×13 mm membrane filter for clarification of aqueous and mild organic solution (Millex- HV, Millipore).
A Centrifugal evaporator (JOUAN RC10.09), coupled to a refrigerated RCT90 Aspirator from Shimadzu (Madrid, Spain), was used to evaporate the extracts of the water samples.
Operating conditions
Standard and water samples: Stock standard solutions (200 mg L-1) were prepared by dissolving pure solid samples in acetonitrile:water (50:50 v/v) for SI and AT, and in methanol:water (50:50 v/v) for DIA, DEA, H-SI and H-AT. Three drops of formic acid were previously added to hydroxyl-triazines to get their complete dissolution.
Working standard solutions were daily prepared by dilution of suitable aliquots from the stock solutions in methanol. All solutions were stored at 4°C in the dark.
Standard water samples were prepared by spiking different amounts (from 0.1 μg L-1) of every herbicide in tap or Milli-Q water samples.
Natural surface water samples were obtained from the 23th aquifer (Albacete, Spain), where they were collected in plastic containers, and directly transferred into 1 L glass topaz bottles. After their collection, these water samples were immediately filtered at laboratory using a 0.45 μm filter to remove the suspended matter, and finally they were kept in the dark at 4°C until their later analysis.
Both tap and natural surface water samples were previously adjusted to 7.0 pH value, before their analysis.
Off-line solid phase extraction and HPLC procedure:
SPE procedure: The extraction and isolation of triazinic herbicides and their degradation products from the spiked tap water and from natural surface water samples, were performed by a SPE process using the already cited 6-mL Bond Elut-ENV SPE-cartridges. Owing to the high level of complexity of the natural surface water matrix, where the analytes under investigation were present at very low levels, the SPE process was used for both pre-concentration and cleaning up stage.
Every step of this extraction procedure was carefully studied and the results were as follows: the cartridge was previously conditioned with 5 mL methanol and 10 mL phosphate buffer 10 mM, pH 7. After that, volumes of water samples (1000 mL) were passed through stationary phase (flow rate <20 mL min-1), then the cartridge was washed with 10 mL phosphate buffer (10 mM, pH 7), and finally the analytes were eluted with methanol (5 mL, three times) into conical glass vessels.
To reach a high enrichment factor in the extractionpreconcentration procedure, the eluted volume (15 mL) was evaporated at 60°C until 200 μL as final volume. Besides, it was necessary to add 100 μL of acetonitrile to the final extract, since it provided a better peak shape and lower baseline noise, reaching so an enrichment factor of 3333. Those final extracts were filtered through a 0.45 μm×13 mm (Millex-HV) filter and immediately injected into the HPLC-DAD equipment.
HPLC-DAD procedure: A Khromasil C18 column as reversed stationary phase for liquid chromatography was selected for the development of analytical separation, whereas a binary gradient elution program combining phosphate buffer 5 mM, pH 7.0 and acetonitrile was chosen as mobile phase.
An elution gradient using a constant flow rate of 1 mL min-1 and an injection volume of 20 μL was used during the analytical separation.
The gradient program profile selected was as follows
| Step | Acetonitrile (%) | Time (min) |
| 1 | 20 | 1 |
| 2 | 20 to 40 | 1 |
| 3 | 40 | 10 |
| 4 | 40 to 60 | 1 |
| 5 | 60 | 1 |
| 6 | 60 to 20 | 1 |
| 7 | 20 | 5 * |
*Getting the initial conditions, reaching so the separation of the six analytes in a running time lower than 14 min, as it can be seen in Figure 1.

Figure 1: HPLC-DAD separation for a standard mixture (10 mg L-1) of hydroxylsimazine (1), deisopropyl-atrazine (2), hydroxyl-atrazine (3), deethyl-atrazine (4), simazine (5) and atrazine (6), obtained in the selected chromatographic conditions, and with the gradient profile also shown.
A wavelength of 215 nm was selected as optimal for detection and quantification of the six herbicides. Nevertheless, on the validation procedure, detection wavelengths were selected at the maximum of spectra for every herbicide, with the purpose to increase the selectivity and decreasing the baseline noise.
Preliminary studies about SPE of s-triazines from natural water samples
As it has been mentioned in the introduction, several works using SPE as isolation procedure and different adsorbents, such as C18 or polymeric ones [10-14], have been published. Currently, innovative material has been successful used with exceptional merit as SPE adsorbents for enrichment of environmental pollutants in water samples [15,16].
In our work, to achieve as much as possible the isolation of s-triazines compounds and their major degradation products from natural water samples, different commercial available cartridges and extraction disks were tested.
Several kinds of cartridges with different polarity, amounts of stationary phases, etc. were compared in order to select the most suitable one to achieve the SPE process. All this information is available as “Supplementary Material”.
Once selected the most suitable cartridge (polarity degree and amount of stationary phase), every step of the SPE procedure, such as the appropriate organic and aqueous solvents, volume for washing stages, samples volume, elution volume and final volume of extracts, was evaluated to achieve a complete extraction. The best recoveries were reached with the optimized procedure already described in Experimental section.
High performance liquid chromatography procedure
The influence of chromatographic parameters like both nature of buffer, pH, ionic strength of mobile phase, percentage of organic modifier, and mobile phase flow rate, was studied on retention times (RT) and resolution between peaks (RS). The selection of these chromatographic parameters was performed attending to reach the best sensitivity and efficiency (N), a good RS between peaks and shorter analysis times.
Preliminary studies were carried out in isocratic mode using different percentages of acetonitrile or methanol in the mobile phase. Acetonitrile was chosen as the organic modifier because it provided higher sensitivity and better peak shape.
Due to the ionization groups of studied triazines (whose chemical structures are shown in Table 1), it was necessary to use a buffer solution as aqueous mobile phase to control the pH. Under knowledge of pKa values of compounds (Table 1), a pH range was studied between 3.0 and 7.0 using phosphate buffers. A 7.0 pH value was selected as optimum because it provided non-ionic forms for the analytes, getting better interaction with the stationary phase, increasing so the RTs, and improving thus the selectivity for the herbicides.
An ionic strength of 5 mM phosphate buffer and a flow rate of 1.0 mL min-1, were chosen as a compromise solution between a good RS (>1.5), higher N, good peak shape and lower analysis times.
On the other hand, a gradient elution program was required to achieve a suitable separation since isocratic mode exhibited too long RTs for SI and AT, as well as RS values lower than 1.5 between H-AT and DEA peaks. The best separation was carried out with the gradient profile already reported in the Experimental section 2.3.2.
A diode array detector was selected taking into account the good spectrophotometric absorption properties (and therefore very good sensitivity) of these aromatic compounds. A wavelength of 215 nm was chosen as optimal one for detection and quantification of the six herbicides.
Figure 1 shows a chromatogram for a standard mixture of the six analytes (10 mg L-1), obtained under the already selected chromatographic conditions. The selected gradient profile is also shown in this figure, which provides a very good resolution between peaks in a run time about 14 min.
Validation off-line SPE HPLC-UV procedure
To assess the reliability of the whole off-line SPE HPLC-DAD procedure, its analytical performance characteristics such as stability, precision, linearity and limits of quantitation among others, were evaluated on extracts obtained from spiked tap water samples. The use of spiked tap water as matrix validation was due to two reasons. The first one was because of tap water matrix was supposed to be free of targeted pesticides, and therefore, we considered it as the most appropriate one to performed precision and accuracy in a reliable way. The other reason was due to the high variability of the different surface water samples from aquifer, and obviously, the inherent difficulty to found any suitable and representative surface water sample of this variety, to carry out a valuable validation study on it. The extracts were obtained by the SPE procedure previously described in Section 2.3.2, which provided an enrichment factor of 3333 for every compound.
Figure 2a presents the off–line SPE-HPLC-DAD separation, corresponding to the extract obtained from a spiked tap water sample (0.30 μg L-1 for every analyte).

Figure 2: a) Off-line SPE HPLC-DAD separation corresponding to an extract from a spiked tap water (0.30 μg-1 L for every herbicide). b) Off-line SPE HPLCDAD screening from natural surface water sample corresponding to sounding 5, where SI, H-AT and DEA were found.
Stability
Although this aspect of the study is often considered to be related to the robustness of the procedure, this factor should be examined at the beginning of the validation process, because it determines the validity of the obtained data in the subsequent tests.
Stability of stock standard solutions was determined by comparing the response factors (concentration/average peak areas) of solutions stored in darkness at 5°C, with those ones of freshly prepared solutions, for triplicate injections each one. All of the studied herbicides were found stable at least for two months (with differences in response factors lower than 0.2%), whereas extracts of spiked tap water were just stable for ten hours.
Precision
The precision of the proposed method has been expressed in terms of Relative Standard Deviation (RSD). Repeatability and intermediate precision were evaluated in accordance with ICH (International Conference on Harmonisation) criteria [31].
To check the repeatability of the chromatographic proposed method, twelve extracts from spiked tap water samples containing 3 μg L-1 of every endocrine-disrupting compound were injected and analyzed. The obtained RSD values were below 3% for peak areas, about 1% for RS and lower than 1% for RTs.
The intermediate precision of the overall off-line SPE HPLC-DAD procedure was evaluated by submitting six spiked tap water samples (3.5 μg L-1 for each compound) to this overall extraction-liquid chromatographic process in triplicate injections in two different days. The Snedecor F-test was used to compare both series of data from the two different days [32]. Significant differences were not observed at a confidence level of 95% and 5 degrees of freedom, since the reached experimental F values were lower than their respective theoretical ones, as it can be seen in Table 2.
| Pesticide | Intermediate precisiona) | LOQb) (ng L-1) | Equationc) | Standard Deviation (SD) SlopeIntercept | σ2d) | Tap Watere) | DeionisedWatere) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Recoveries | Recoveries | ||||||||||
| Fexpfor Area | Fexpfor RS | ||||||||||
| % | SDf) | % | SDf) | ||||||||
| H-SI DIA H-AT DEA SI AT |
1.18 1.39 1.33 1.71 1.05 1.28 |
2.92 4.46 3.12 3.20 1.47 4.93 |
96 88 90 81 95 100 |
y = 120205 CHSI - 23728 y = 143618 CDIA - 37688 y = 108686 CHAT - 9760 y = 147826 CDEA - 31214 y = 70148 CSI - 16736 y = 112109 CAT - 5123 |
14003 36842 18657 26269 13935 25808 |
18197 14189 3569 2898 8061 1970 |
0.9985 0.9929 0.9968 0.9965 0.9957 0.9943 |
71.50 86.32 77.40 91.67 74.47 71.20 |
3.20 5.63 1.13 4.22 4.15 3.82 |
69.19 89.24 71.62 94.29 78.00 74.14 |
2.06 2.65 0.76 2.99 3.72 0.56 |
a)F(5, 5) theorical=5.05
b)LOQ including enrichment factor (3333)
c)Peak area (y, no units) versus concentrations (μg L-1).
d)Regression coefficients
e)Mean of recoveries in spiked tap and deionized water samples at three different levels in triplicate.
f)Mean of standard deviations (SD) of recoveries in spiked tap and deionized water samples at three different levels in triplicate
Table 2: Analytical performance characteristics for the developed off- line SPE- HPLC-DAD method.
LODs and LOQs
Limits of detection and quantification (LODs and LOQs) were estimated upon IUPAC criteria, by evaluating the baseline noise on six blank tap water extracts using the maximal sensitivity allowed by the system, over a period ten times the peak width, around the specific retention times for every herbicide. They were found values between 90 and 100 μg L-1 for LODs and from 300 to 350 μg L-1 for LOQs. These LOQs values were subsequently validated by the analysis of five different blank tap water extracts, spiked with amounts of each analyte corresponding to their respective LOQs. The relative errors obtained in the verification of these LOQs were lower than 15% in all of the cases.
It is important to emphasize that according to the preconcentration factor reached in the SPE step, the LODs and LOQs allowed in natural water samples could be 3333 times lower than the HPLC-DAD calculated values (Table 2). Consequently, as it be can see in Table 2, the LOQs reached for studied herbicides (between 81 and 100 ng L-1), are from ten to thirty fold lower than the maximal allowed amounts for natural surface waters (1 and 3 μg L-1 as alert and alarm levels), according to European Union directives [3].
Linearity
The linear behaviour was determined from triplicate injections of spiked tap water samples, previously submitted to the SPE treatment, at six different concentration levels between 0.1 and 1.0 μg L-1 for every compound. The satisfactory linear regression equations, their standard deviations for slope and intercept and their regression coefficients are also summarized in Table 2. The obtained results can indicate that SI, AT, DIA, DEA, H-SI and H-AT responses are linear over the studied concentration range.
However, according to the Analytical Methods Committee (AMC) [33], a regression coefficient close to unity is not necessarily the outcome of a linear relationship, and as consequence, the lack of fit test was carried out by plotting the residuals (distances of the experimental points from the fitted regression lines) against concentration for every analyte. If, there is not lack of fit, the plot will look like a random sample from a normal distribution with zero-mean that is the calibration results inherently linear. This one was the observed situation on applying this test to our calibration graphs, and therefore the linearity of these relationships was thus confirmed.
Accuracy
To test the accuracy of the proposed procedure, three tap and three deionized water samples spiked with the six herbicides in variable amounts between 0.1-2 μg L-1 (in triplicate), were submitted to the off-line SPE HPLC-UV procedure. The results (average recoveries), summarized in Table 2, were between 85-95% for DIA and DEA and between 70-80% for the other herbicides. In this table, it can also be noted that recoveries are quite similar for deionised and tap water samples. Therefore, we can say that the proposed SPE procedure reach to remove the presence of salts, suspension and dissolving matter in these tap water samples.
Robustness
Robustness evaluates the influence of small changes on the internal factors of an analytical procedure, providing an indication of its reliability during normal usage [34]. The purpose of a robustness test is to identify possible sources of errors, when changes occur in the specified method conditions [35-38]. In this paper we have tested the influence of small variations on internal parameters of the method, whose influence has been studied at three different levels (Table 3).
| Factor | Units | Limits | Level(-1) | Level (+1) | Nominal(0) |
|---|---|---|---|---|---|
| A. pH phosphate buffer | ± 0.5 | 6.5 | 7.5 | 7 | |
| B. Buffer concentration | mM | ± 2 | 3 | 7 | 5 |
| C. Flow rate | mL min-1 | ± 0.2 | 1.2 | 0.8 | 1.00 |
| D. Initial % solvent B (step1) | % | ± 2 | 18 | 22 | 20 |
| E. First gradient program time (step 2) | Min | ± 0.2 | 0.8 | 1.2 | 1 |
| F. % solvent B (step 3) | % | ± 2 | 38 | 42 | 40 |
| G. Second held time (step 3) | Min | ± 1 | 9 | 11 | 10 |
Level 0: optimal values
Level +1: upper values
Level -1: lower values
Table 3: Variables selected as factors and values chosen as levels.
The Plackett-Burman fractional factorial model, based on balanced incomplete blocks has been employed for this evaluation, using a design for seven factors and fifteen experiments (N=15) [39].
The mean effect of each variable is the average difference between observations made at the extreme levels and those made at the optimal one. Mean effects and standard errors (DA, DB, DC...) were calculated using the procedures described by Youden and Steiner [34].
Results of the levels variation effects were checked on the most critical chromatographic responses of the method: resolution between H-SI and DIA, between H-AT and DEA, and AT peak area efficacy. Taking into account these obtained deviations, the proposed method has proved to be robust for all of the variations tested in this study, since in all of the cases for the operating factors (A-G), on every studied chromatographic response, the following rule was observed:
being
Figure 3 shows the effects of negative (-1) and positive (+1) levels for the seven selected factors on RS between H-AT and DEA, where as it can be seen the most critical factors resulted to be the % acetonitrile at positive level in the four step, and also the influence of buffer concentration at negative level. Anyway, the proposed analytical method has proved to be enough robust, when the influence of these seven factors was evaluated on this critical chromatographic response.

Figure 3: Variation effects of the levels (-1) and (+1) for the seven selected operating factors on RS between H-AT and DEA.
Peak homogeneity
It is useful to investigate the purity of separated peaks because comigration of peaks is also possible in HPLC, as in any other separation technique. Several techniques for validating peak purity have been proposed in the literature [40,41].
In our work two techniques have been used to validate the peak purity of the compounds under study.
One of them, called “normalization and comparison of spectra at different peak sections”, where the absorption spectra acquired at the apex, at the ascending and at the descending parts of the corresponding peak for every herbicide present in each sample, were compared.
The second one, named “absorbance at two wavelengths” where a compound can be define as “pure”, when the molar absorptivity (A) at a wavelength λ1 is directly proportional to that one at any other wavelength λ2, that means, A (λ1)=KA (λ2); where K characterizes the pure compound at the selected wavelength. The absorbance ratio for a pure compound is constant. A plot of this ratio against RTs will give a square wave function of K amplitude value. Any co-eluting impurity peak leads to a deviation from the flat ratio plot. The wavelengths have to be selected at the highest absorbance difference. An obtained square wave function demonstrates the purity of the chromatographic peaks. In all of our applications analyzed by the proposed method, similar results to describe above were obtained. Both methods were satisfactorily applied to check the lack of interferences from the matrix in natural surface water samples from aquifer, on the peaks assigned as focused compounds.
Application to natural water samples
Natural surface water samples from different soundings at several locations in the 23th aquifer (Albacete, Spain), have been analyzed using the proposed off-line SPE HPLC-DAD procedure. In Table 4 has been summarized the results obtained for quantitative analyses.
| Sounding | H-SI | DIA | H-AT (μgl-1±SD | DEA | SI | AT |
|---|---|---|---|---|---|---|
| 1 | 0.24±0.06 | n.d | 0.31±0.09 | 0.12±0.03 | n.d | n.d |
| 2 | 0.21±0.07 | n.d | 0.17±0.05 | 0.12±0.03 | n.d | n.d |
| 5 | n.d | n.d | 0.28±0.05 | 0.24±0.08 | 0.20±0.06 | n.q |
| 6 | n.d | 0.12±0.03 | n.q | n.q | n.d | 0.13±0.03 |
| 7 | n.d | n.q | n.q | n.d | n.d | n.d |
| Location 1 | n.q | n.d | n.q | n.d | n.d | n.d |
| Location 2 | n.d | n.d | 0.23±0.05 | n.d | n.d | n.d |
| Location 3 | n.q | n.d | 0.10±0.03 | n.d | n.d | n.d |
n.d=no detected
n.q=no quantificated,
Table 4: Concentrations (μg L-1) and Standard Desviation (n=3) found for every herbicide in the different soundings from the 23rd aquifer (Albacete, Spain).
As it can be observed, AT has been detected in two samples, whereas SI was just quantitated in one sounding corresponding to the fifth one. The studied degradation products H-SI, H-AT, DIA and DEA were found between concentration values of 0.10 and 0.31 μg L-1 in the different soundings (Table 4). H-AT was present in all of the analyzed samples, whereas DIA was found in just two of them. These results support the presence of main degradation products of parents SI and AT, proving so the great importance to include them as aim of study for the analysis of environmental samples as natural surface water samples.
The Figure 2b shows an off-line SPE HPLC-DAD screening from sounding 5, where amounts of SI and two degradation products (DEA an H-AT) were found.
The specificity of targeted analytes in the natural surface water samples, was checked by the two methods already described in “peak homogeneity” section, in order to perform a reliable assignment of quantified analytes.
Previously the authors proposed an off-line SPE GC-MS-SIM method, to perform the qualitative and quantitative determination of other s-triazine herbicides and related degradation products, where AT, SI, DIA and DEA were satisfactorily analyzed [23]. However, this method did not permit to determine OH-triazine degradation products, H-AT and H-SI, without a previous derivatization step. For this reason, it was necessary to develop an alternative and simple method, which could do it with LOQs at similar ranges, allowing us in this way to quantify these hydroxylated degradation products, which are very often found at relevant levels in the natural surface water samples, as we can see in Table 4.
In this work, a sensitive, easy and simple chromatographic method has been described for the separation and quantification of endocrinedisrupting chemicals SI, AT and their major degradation products as hydroxylated and dealkylated ones, H-SI, H-AT and DIA and DEA, respectively. A SPE procedure for the isolation and preconcentration of these analytes from water matrix was developed. The SPE procedure was extensively evaluated to obtain a high enrichment factor, for achieving a complete extraction and reaching a sensitivity increase. The instrumental conditions for this off-line SPE HPLC-DAD method were established for determination at trace levels of these six herbicides, widely found in Spain, in natural surface and ground water samples.
The reliability of the off-line SPE HPLC-DAD method was successfully proved by a validation procedure including stability of solutions, precision, linearity, determination and quantification limits, accuracy, statistical robustness evaluation and specificity studies.
Since our method provides enough low LOQs to quantify all of the analytes (from ten to thirty fold lower) than the relevant maximal amounts permitted in natural surface waters (1 and 3 μg L-1 as alert and alarm levels), upon European Union directives [3], it was also applied to qualitative and quantitative analysis of endocrine-disrupting herbicides in different natural surface water samples from the 23rd aquifer.
The results confirmed mainly the presence of degradation products such as H-AT, H-SI and DEA, at concentration levels of μg L-1, proving so the importance to include the mentioned dealkylated and hydroxylated-triazines as targeted analytes. Among the different soundings, AT, SI and DIA were quantified in just one sample (different among them), whereas H-AT was present in all of the analyzed surface waters from aquifer. The degradation products H-SI, H-AT, DIA and DEA were found between concentration values of 0.10 and 0.31 μg L-1.
As added advantage, this method allows the analysis of hydroxyldegradation products, analytes which are difficult to analyse by GC methods without previous derivatization, but nevertheless, they are very common in natural surface waters from aquifer, and consequently, they are very important matter of analysis, as we can see from the results shown in Table 4.
Another main value of our method remains on its proved reliability, even for the analysis of the degradation products, whose study has been reported by few papers until now, despite their herbicide activity too. This is in opposite to other more recent works previously published, like the use of MWCN as sorbent in SPE process, or either the use of back pressure monolithic columns. Both of them have been used until now, just for the analysis of the parent compounds, but not for their degradation products yet. The results confirmed mainly the presence of degradation products such as H-AT, H-SI and DEA, at concentration levels of μg L-1, proving so the importance to include the mentioned dealkylated and hydroxylated-triazines as targeted analytes.
The authors would like to thank“Convenio específico de colaboración entre Consejería de Obras Públicas de la Junta de Comunidades de Castilla –La Mancha y la Universidad de Castilla la Mancha en materia de calidad de aguas” and“Centro Regional de Estudios del Agua (CREA)”.
Several types of cartridges, changing polarity of column, amounts of stationary phases, i.e. C18 cartridges (500, 1000 and 2000 mg), styrene divinylbenzene cartridges (500 and 1000 mg) and C18 disks (diameter 47 mm) were compared. It was observed the co-polymer styrene divinylbenzene-1000 mg cartridges provided higher recoveries (70-90%), than the recoveries (<50%) obtained using reversed phases (C18 cartridge or disks). This can be probably due to the non-ionic character of studied compounds, so this stationary phase was selected for later studies.