Journal of Chromatography & Separation Techniques
Open Access
ISSN: 2157-7064
ISSN: 2157-7064
Research Article - (2015) Volume 6, Issue 5
Risk profile and uncertainty estimation are the two major and important parameter that need to be carried out during the development of pharmaceutical process, to obtain reliable results. The conventional method validation schedule needs to be improvised so as to certify extraordinary method reliability to measure quality feature of a drug product. Risk profile assessment, expanded uncertainty and combined standard uncertainty in the analysis of cilostazol and telmisartan in combined tablet dosage form were studied in this research work. RP-HPLC method was validated in our laboratory as per ICH guideline and risk profile assessment has been outlined including uncertainty estimation using the cause-effect approach. In the course of validation, the calibration model found to be impregnable when encountered with lack of fit test and Levene’s test. In uncertainty major contribution is due to sample concentration and mass. The proposed research work clearly demonstrate the application of theoretical concept of calibration model tests, relative bias, risk profile and uncertainty in the methods used for analysis in drug discovery process.
Keywords: Cilostazol (CLZ); Telmisartan (TLM); RP-HPLC; Risk profile; Relative bias; Combined standard uncertainty; Expanded uncertainty
Decisions are made by measurement. When we use number to make decision every time we run with the risk of making fault, because all number are more or less reliable. Since every measurement is suspected, it is necessary to know how and why it is so. So to avoid this risk of making mistake with every measurement and to clear worries associated with measurement recently a new approach has been familiarized known as measurement of uncertainties in analysis of sample. The quality of method is expressed in terms of its uncertainty and assessment of uncertainty becomes key parameter for method validation to get certification [1,2]. Analytical method for evaluation of drug component in pharmaceutical formulation has been introduced and available worldwide. But the validation by total error approach and quantification of causes of uncertainties in this developed and published method has been omitted. In this study simple method for quantification of uncertainty components and combined standard uncertainty (CSU) is presented by assessing this computation for RPHPLC measurement of cilostazol and telmisartan in tablet dosage form. Some article recommends the conventional estimation of analytical measurement and uncertainty as well. For perfect and complete study of uncertainty components, a new RP-HPLC method for cilostazol and telmisartan in combined tablet dosage form has been established and validated as per the ICH guideline [3,4].
A simple, rapid, accurate, precise, reliable and economical RPHPLC method with UV detection was optimized, developed and validated as per ICH-Q2 guideline for the simultaneous estimation of cilostazol (CLZ) and telmisartan (TLM) in tablet dosage form. The retention behavior of CLZ and TLM as a function of mobile phase pH, composition and flow rate was inspected. Separation was developed on a reverse-phase C18 column (250 mm × 4.6 mm i.d, 5 μ particle size), using a mobile phase consisting of Acetonitrile: Phosphate Buffer (pH-3.5 adjusted with Orthophosphoric acid) in the ratio of a 60:40 (%v/v) at a flow rate of 1.0 ml/min with UV detection at 255 nm within 10 min with retention time of 4.23 and 6.34 for CLZ and TLM respectively. The standard curves were linear over the concentration range of 12-32 μg/mL and 3-8 μg/mL for CLZ and TLM. The developed method was validated in terms of accuracy, precision, and linearity, limit of detection and limit of quantification. From the validation outcomes it was established that proposed method can be used for the approximation of both drugs in combined pharmaceutical tablet dosage form.
Cilostazol (CLZ) (Figure 1) is chemically known as 6-[4-(1-cyclohexyl-1H-tetrazol-5-y1) butoxy]-3,4-dihydro-2 (1H) – quinolinone and is a quinolinone derivative that inhibits cellular phosphodiesterase III, and is used for the inhibition of platelet aggregation and as a vasodilator [5-6]. Telmisartan (TLM) (Figure 2) is chemically known as 4'-([4-methyl-6-(1-methyl-lH-benzimidazol- 2yl)-2-propyl-lH-benzimidazol-l-yl]methyl}-2-biphenylcarboxylic acid. Telmisartan is a new angiotensin II receptor antagonist for the treatment of essential hypertension and useful in the treatment of mild to moderate hypertension, well tolerated with a lower incidence of cough than ACE inhibitors [7,8]. Several method for determination of cilostazol and telmisartan individual and in combination with other drug has been reported in the past, such as spectrophotometric [9-12], high-performance liquid chromatography (HPLC) [13-15]. Although the RP-HPLC method for simultaneous estimation of cilostazol and telmisartan is already developed but this method include the uncertainty measurement and [16-21] risk profiling using total error approach and developed method is more economical as compare to previous published method [22].

Figure 1: Structure of cilostazol.

Figure 2: Structure of telmisartan.
The present research paper describes a validated RP-HPLC method for quantification of cilostazol and telmisartan in combined dosage formulation with total error approach and uncertainty measurement.
HPLC Instrumentation
Chromatography was performed on Shimadzu (Shimadzu Corporation, Kyoto, Japan) chromatographic system equipped with Shimadzu LC-20AT pump and Shimadzu SPD-20AV absorbance detector. Samples were injected through a Rheodyne 7725 injector valve with fixed loop at 20 μl. Data acquisition and integration was performed using Spinchrome software (Spincho biotech, Vadodara). The chromatographic elution of analytes was obtained by using Kromasil C18 5 μm (250 × 4.6) mm column at flow rate of 1 ml/min was used.
Reagents and chemicals
Telmisartan was produced as gift sample from Alembic pharmaceutical, Vadodara and cilostazol was purchased from Swapnroop drug Pvt.. Ltd Bombay. HPLC grade Methanol, Acetonitrile and Ortho Phosphoric Acid were supplied by Spectrochem Pvt. ltd, Mumbai. Water used throughout the experiment was Purified HPLC grade water obtained by filtering double distilled water through nylon filter paper 0.2 μm pore size and 47 diameters (Pall Life sciences, Mumbai, India).
Preparation of mobile phase
Phosphate Buffer of pH 3.5 (pH adjusted with Ortho phosphoric acid ) was prepared and this phosphate buffer was filtered through 0.20 μm filter paper and then proper mixing of Acetonitrile and Phosphate Buffer (60:40) was done. Then the prepared mixture were sonicated for 5 min in ultrasonic bath, and then used as mobile phase.
Preparation of standard solutions of CLZ and TLM
Stock solution of (1000 μg mL-1) for both cilostazol and telmisartan was prepared by accurately weighing 25 mg of both drugs in 25 ml volumetric flask and then diluted with Methanolup to the mark. From this stock 10 ml solution was withdrawn and transferred to 100 ml of volumetric flask and then diluted up to the mark with Mobile phase to get working standard solution of (100 μg mL-1) of cilostazol and telmisartan.
Preparation of calibration curve
From the working standard solution of cilostazol (100 μg mL- 1) aliquots ranging from 1.2 ml to 3.2 ml were taken, in 10 ml volumetric flask and diluted to volume with mobile phase to give final concentrations of 12, 16, 20, 24, 28, 32 μg mL-1 of CLZ. And aliquots ranging from 0.3 ml to 0.8 ml were taken, from the working standard telmisartan (100 μg mL-1) in 10 ml volumetric flask and diluted to volume with mobile phase to give final concentrations of 3, 4, 5,6 7, 8 μg mL-1 of TLM.
Injections of 20 μl were made for each concentration and chromatogram was obtained under the condition described in. Calibration graph was constructed by plotting peak area versus concentration of each drug and the regression equation was calculated.
Selection of common solvent
CLZ and TLM were found to be soluble in methanol and hence, they were first dissolved in methanol and then, to avoid interference of methanol in chromatograms, the dilutions were made with mobile phase.
Selection of detection wavelength
The detection wavelength should be the one where both the drugs show considerable absorbance for the purpose of obtaining good sensitivity. Both the drugs are having appreciable absorbance at 255 nm hence 255 nm selected for detection.
Selection and optimization of chromatographic conditions
To optimize the chromatographic conditions, the effect of chromatographic variables such as composition of mobile phase, pH of mobile phase and flow rate were studied. The resulting chromatograms were recorded and the chromatographic parameters such as capacity factor, asymmetric factor, resolution and theoretical plates were calculated plates were calculated. The conditions that gave the best resolution, symmetry and theoretical plate were selected for estimation. Several mobile phases with different pH and ratio were tried and good and well resolved peak was obtained in Acetonitrile: Phosphate Buffer (60:40 pH 3.5).
Validation procedure using total error approach
Current ad-hoc approaches to method validation are inconsistent with ensuring method suitability. A total error approach based on the use of two-sided b-content tolerance intervals was developed. The total error approach offers a formal statistical framework for assessing analytical method performance. The approach is consistent with the concept of method suitability and controls the risk of incorrectly accepting unsuitable analytical methods. Risk profiling is a process for finding the optimal level of risk for developed method and risk associated with method. The present method was validated as per ICH guideline [3,4] and ISO guideline which were grounded upon “total error” approach [1]. In this approach “total error” was estimated by merging the systemic error and random error to recognize the difference between observed and true value. In the proposed method sensitivity of the method and effect of sample matrix were also studied. The selectivity of the studied method was investigated by comparing chromatogram of blank without CLZ and TLM, blank mobile phase and sample with CLZ and TLM and sample of formulation. Response function in proposed method three sets of calibration curve were plotted between area and different concentration of CLZ and TLM and on these three different series regression analysis was performed and series with best coefficient of determination was selected and the selected series has been further diagnosed by Levene's test (Table 1) and standardized residual plot (Figures 3 and 4). Trueness of calibration curve was calculated by back calculation of concentration to justify the calibration line. The result of trueness was expressed in terms of absolute and relative bias (Tables 2.1 and 2.2). In order to confirm the reproducibility of standard precision at two different levels were studied, first one is repeatability under same operating condition over short time interval and second is intermediate precision assessed on different days. The precision result expressed in terms of % relative standard deviation (RSD) (Tables 3.1 and 3.2) (Figures 5 and 6). Relative and absolute precision at both level were calculated with 95% Upper confidence limit (Tables 3.1 and 3.2). The recovery study which is the most critical parameter in method validation requires an extra precaution during study and interpretation of recovery results. Therefore, the results of accuracy studies were interpreted and represented in the β- expectation tolerance limits (Tables 4.1 and 4.2). In addition to this parameter, risk profile has also been studied to know the future application of the method. Linearity profile was also studied demonstrate the relationship between nominal and observed concentration in matrix and furthermore, residual plot was generated to know the outliers in the determination of CLZ and TLM in sample matrix (Figure 7). Limit of detection and quantification represents the sensitivity of the method which has been calculated as per ICH guidelines. Limit of detection LOD and limit of quantification are two important parameter which show the application of method in quantification and detection of different sample. These are calculated according to the procedure mentioned in the ICH guideline [4,5].
| Source | SS | df | MS | Fcalc | Fcrit,95% | p-value | |
|---|---|---|---|---|---|---|---|
| CLZ | Model | 12.88 | 5 | 2.576 | 2.299 | 2.409 | 0.05947 |
| Error | 53.77 | 48 | 1.12 | ||||
| TLM | Model | 5.213 | 5 | 1.043 | 4.324 | 2.409 | 0.002501 |
| Error | 11.57 | 48 | 0.2411 |
Table 1: Evaluation of homogeneity of variances (Levene's test) for CLZ and TLM.
| Concentration level (mcg/ml) | Mean introduced concentration (mcg/ml) | Mean Back calculated concentration (mcg/ml) | Absolute bias (mcg/ml) | Relative Bias(%) | Recovery (%) | 95% Confidence Interval of Recovery (%) |
|---|---|---|---|---|---|---|
| 1.0 | 12.00 | 11.63 | -0.3668 | -3.057 | 96.94 | [ 96.27 , 97.61] |
| 2.0 | 16.00 | 16.32 | 0.3215 | 2.009 | 102.0 | [ 101.7 , 102.4] |
| 3.0 | 20.00 | 19.99 | -0.008377 | -0.04188 | 99.96 | [ 99.69 , 100.2] |
| 4.0 | 24.00 | 23.99 | -0.005115 | -0.02131 | 99.98 | [ 99.77 , 100.2] |
| 5.0 | 28.00 | 28.58 | 0.5838 | 2.085 | 102.1 | [ 101.8 , 102.4] |
| 6.0 | 32.00 | 31.48 | -0.5249 | -1.640 | 98.36 | [ 98.10 , 98.62] |
Table 2.1: Result of Trueness in terms of relative bias (%) for CLZ.
| Concentration level(mcg/ml) | Mean introduced concentration(mcg/ml) | Mean back calculated concentration(mcg/ml) | Absolute bias(mcg/ml) | Relativebias (%) | Recovery (%) | 95%Confidenceinterval ofrecovery(%) |
|---|---|---|---|---|---|---|
| 1.0 | 3.000 | 2.947 | -0.05291 | -1.764 | 98.24 | (97.60, 98.87) |
| 2.0 | 4.000 | 4.059 | 0.05864 | 1.466 | 101.5 | (100.8, 102.2) |
| 3.0 | 5.000 | 5.009 | 0.009405 | 0.1881 | 100.2 | (99.37, 101.0) |
| 4.0 | 6.000 | 5.956 | -0.04397 | -0.7328 | 99.27 | (98.56, 99.98) |
| 5.0 | 7.000 | 7.090 | 0.08972 | 1.282 | 101.3 | (101.0, 101.6) |
| 6.0 | 8.000 | 7.939 | -0.06088 | -0.7610 | 99.24 | (99.03, 99.45) |
Table 2.2: Result of Trueness in terms of relative bias (%) for TLM.
| Nominalconc (mcg/ml) | Repeatability (RSD%) | Intermediateprecision (RSD%) | Repeatability (SD-mcg/ml) | Intermediate precision (SD - mcg/ml) | 95% Upper confidence limit repeatability (SD -mcg/ml) | 95% Upper confidencelimit intermediate precision (SD - mcg/ml) |
|---|---|---|---|---|---|---|
| 12.00 | 0.8732 | 0.8732 | 0.1048 | 0.1048 | 0.1793 | 0.1793 |
| 16.00 | 0.4602 | 0.4602 | 0.07364 | 0.07364 | 0.1260 | 0.1260 |
| 20.00 | 0.3487 | 0.3487 | 0.06973 | 0.06973 | 0.1193 | 0.1193 |
| 24.00 | 0.2665 | 0.2665 | 0.06397 | 0.06397 | 0.1094 | 0.1094 |
| 28.00 | 0.4125 | 0.4125 | 0.1155 | 0.1155 | 0.1976 | 0.1976 |
| 32.00 | 0.3444 | 0.3444 | 0.1102 | 0.1102 | 0.1886 | 0.1886 |
Table 3.1: Result of relative and absolute Intermediate Precision and Repeatability in terms of (%RSD) CLZ.
| Nominalconc (mcg/ml) | Repeatability (RSD%) | Intermediate precision (RSD%) | Repeatability (SD-mcg/ml) | Intermediateprecision (SD –mcg/ml) | 95% Upper confidence limit repeatability (SD –mcg/ml) | 95% Upper confidencelimit intermediate precision (SD - mcg/ml) |
|---|---|---|---|---|---|---|
| 3.000 | 0.8265 | 0.8265 | 0.02479 | 0.02479 | 0.04242 | 0.04242 |
| 4.000 | 0.8951 | 0.8951 | 0.03580 | 0.03580 | 0.06126 | 0.06126 |
| 5.000 | 1.069 | 1.069 | 0.05346 | 0.05346 | 0.09147 | 0.09147 |
| 6.000 | 0.9230 | 0.9230 | 0.05538 | 0.05538 | 0.09475 | 0.09475 |
| 7.000 | 0.4117 | 0.4117 | 0.02882 | 0.02882 | 0.04930 | 0.04930 |
| 8.000 | 0.2701 | 0.2701 | 0.02161 | 0.02161 | 0.03697 | 0.03697 |
Table 3.2: Result of relative and absolute intermediate precision and repeatability in terms of (%RSD) TLM.
| Concentration level (mcg/ml) | Meanintroduced concentration (mcg/ml) | Beta-expectation tolerance limits (mcg/ml) | Relative beta-expectation tolerance limits (%) | Risk2 (%) |
|---|---|---|---|---|
| 1.0 | 12.00 | (11.38, 11.89) | (-5.193, -0.9208) | 3.454 |
| 2.0 | 16.00 | (16.14, 16.50) | (0.8831, 3.135) | 0.01566 |
| 3.0 | 20.00 | (19.82, 20.16) | (-0.8949, 0.8111) | 0.0001150 |
| 4.0 | 24.00 | (23.84, 24.15) | (-0.6734, 0.6307) | 0.00001532 |
| 5.0 | 28.00 | (28.30, 28.87) | (1.076, 3.094) | 0.008976 |
| 6.0 | 32.00 | (31.21, 31.74) | (-2.483, -0.7977) | 0.0009638 |
Table 4.1: Method accuracy obtained by considering linear regression for cilostazol.
| Concentrationlevel (mcg/ml) | Mean introduced concentration (mcg/ml) | Beta-expectation tolerancelimits (mcg/ml) | Relative beta-expectation tolerance limits (%) | Risk2 (%) |
|---|---|---|---|---|
| 1.0 | 3.000 | (2.886, 3.008) | (-3.786, 0.2582) | 0.3188 |
| 2.0 | 4.000 | (3.971, 4.146) | (-0.7237, 3.656) | 0.3098 |
| 3.0 | 5.000 | (4.879, 5.140) | (-2.428, 2.804) | 0.2442 |
| 4.0 | 6.000 | (5.821, 6.092) | (-2.991, 1.525) | 0.1481 |
| 5.0 | 7.000 | (7.019, 7.160) | (0.2745, 2.289) | 0.001690 |
| 6.0 | 8.000 | (7.886, 7.992) | (-1.422, -0.1002) | 0.00003217 |
Table 4.2: Method accuracy obtained by considering linear regression for telmisartan.

Figure 3: Standardized residuals plot for cilostazol.

Figure 4: Standardized residuals plot for telmisartan.

Figure 5: Uncertainty profile for cilostazol determination.

Figure 6: Uncertainty profile for telmisartan determination.

Figure 7: Linearity, overlain chromatogram of CLZ (12-28 μg/ml) and TLM (3.0-8.0 μg/ml).
Limit of detection (LOD) and limit of quantification (LOQ) are two important parameter which show the application of method in quantification and detection of different sample. These are calculated according to the procedure mentioned in the ICH guideline [4,5].
Cause-effect diagram:
Even though estimation method was validated as per guidelines but still doubt was there in result as during the validation of method small influence which can affect the results has not been studied, Such as error during sample weighing, discharge of volumetric flask etc. Therefore, to overwhelm such doubt during result collation were clarified by estimation of uncertainty in result obtained from validation. The protocol for uncertainty estimation starts with identification of sources of uncertainty. The best way of listing uncertainty sources is to use the cause-effect diagram plan, as it outlines the sources connection to each other demonstrating their impact on the result. Thus a cause-effect diagram was assembled as presented in Figure 8. The parameter taken in consideration was volume of volumetric flask V10, and mass of sample, recovery of method Rm and precision of method. These all parameter contribute to uncertainty in the interpreted results. This diagram also helps in resolving any repeatability of component in uncertainty. The parameter comes in consideration after constructing cause-effect diagram was illustrated in Equation 1.
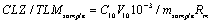 (1)
(1)

Figure 8: Cause and effect diagram.
Where, CLZsample and TLMsample, CLZ and TLM quantity in (mol/ kg); C10, CLZ and TLM concentration in 10 mL volumetric flask (M); V10 volume of 10 mL volumetric flask (mL); msample, CLZ and TLM sample mass taken (kg); Rm, Recovery of method.
These identified sources were quantified and their discrete effect of on inclusive uncertainty was assembled as CSU and EU.
Individual parameter showing effect on overall uncertainty
Liberation of solution from volumetric flask:
The uncertainty due to liberation of volumetric flask was evaluated by performing experiment involving filling up and weighing of 10 mL volumetric flask with standard solution for 10 times.
Mass (msample): Difference between weighing glass with and without the sample provide the sample mass.
Concentration, C10: The uncertainty in concentration of drug obtained from calibration curve is expressed as uncertainty due to concentration C10. This is estimated using Equation 2.
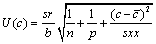 (2)
(2)
Where 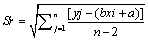 (3)
(3)
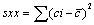 (4)
(4)
Sr standard deviation of residual; n, number of measurement used for calibration curve; p, number of measurement used to obtain concentration of sample; c, concentration in sample (M); c, average of standard solution (M); Yj, response obtained from the measurement; j, index for number of measurement made in order to obtain the calibration curve; i, index for number of solution for calibration; b, slope of calibration curve (L mol-1 ); a, calibration curve intercept;
Recovery of method:
Uncertainty associated with recovery of method was evaluated using Equation 5 and it depends upon spiked and recovered concentration of standard in sample matrix.
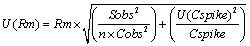 (5)
(5)
Where, Cobs, mean of concentration observed from replicate analysis of spiked sample; Cspike, nominal concentration of drug in spiked sample. Sobs, means standard deviation of result from the replicate analyses of spiked sample; n, number of replicates; U (Cspike), standard uncertainty in concentration of spiked sample.
Precision (P):
During the validation of method precision studies were carried out. In this study repeatability and variability associated with the measurement were included in overall precision uncertainty estimation.
Validation parameters
In this method calibration curves from the response of different concentration were prepared using linear regression model. The four different sets were prepared for response function studies with range of 12-32 μg/ml for CLZ and 3-8 μg/ml for TLM, from their regression analysis studies series 3 show the best results with coefficient of determination r2 0.997 and 0.9986 for CLZ and TLM respectively, so this series was selected for further competition for validation and sample analysis. Moreover, the selected series and regression model was diagnosed and confirmed using lack of fit test (LOF). The p-value were calculated and found to be greater than 0.05 and further to demonstrate that no outliers were found in calibration curve standard residual plot were also plotted as represented in Figures 3 and 4. As the model was established now in order to authenticate the regression equation back calculation were done and linear plot using absolute β-expectation limit was constructed between nominal and back calculated concentration which showing the 0.9968 and 0.9984 for CLZ and TLM respectively and confirming the authenticity of regression equation. Trueness of method justified by calculation of % relative bias which was found to be limited between [-3.057 - 2.085] for CLZ and [1.282- 1.764] for TLM as illustrated in Tables 2.1 and 2.2 from which it has been concluded that the trueness of method is adequate. The method precision and reproducibility was authenticated by result obtained from precision studies which were found to be <2% in terms of RSD for both repeatability and intermediate level as illustrated with 95% confidence upper limit in Tables 3.1 and 3.2. After the confirmation of accuracy of all the parameters related to the system and developed method, sample matrix was incorporated in validation process which includes recovery studies. Recovery studies were carried out using standard addition method in sample matrix. These recovery studies receipts into account total error of test result and is represented by the β-expectation tolerance limit. The result of accuracy studies has been illustrated in Table.4.1 and 4.2. The β-expectation tolerance limit was also found to be in the acceptance as accuracy profile illustrated in Figures 9-12. Further, these recovery studies of model was justified by plotting risk profile keeping maximum risk level at 5.0% from which it was concluded that risk of outliers are within limits and in future analysis of the sample using this developed and validated method will fall within range. The results of LOD show that this method is sensitive enough to analyze marketed formulations; LOD was found to be 1.232 and 0.2174 for CLZ and TLM respectively.

Figure 9: Accuracy profile of cilostazol obtained by considering linear regression the plain red line is the relative bias, the dashed lines are the β-expectation tolerance limits and the dotted curves represent the acceptance limits.

Figure 10: Accuracy profile of telmisartan obtained by considering linear regression the plain red line is the relative bias, the dashed lines are the β-expectation tolerance limits and the dotted curves represent the acceptance limits.

Figure 11: Risk profile obtained by considering linear regression cilostazol, the dotted line represents the maximum risk level chosen: 5.0%.

Figure 12: Risk profile obtained by considering linear regression telmisartan, the dotted line represents the maximum risk level chosen: 5.0%.
Analysis of formulation
It is evident from the aforementioned results that proposed method gave satisfactory results with the CLZ and TLM in bulk drug. The dosage forms were subjected to analysis for their content of active drug material by the proposed method. The percentage purity for tablet was found to be 102.475% for CLZ and 99.50% for TLM (Table 5). It is evident from the above mentioned results that proposed method is applicable to the analysis of drug in its bulk drug as well as synthetic forms with comparable analytical performance.
| Formulation: Synthetic mixture | ||
|---|---|---|
| Drug | Composition of mixture | %Drug found*± SD |
| Cilostazol | 40 mg | 102.475±0.1150 |
| Telmisartan | 10 mg | 99.50±0.0709 |
Table 5: Application of the developed method in synthetic mixture.
Measurement of uncertainty
Once uncertainty sources have been identified, they were evaluated and their magnitude was determined. In order to assure the traceability for uncertainty results all the computation were done in International System of Unit as concentration in M and weight in kg.
Uncertainty of volumetric flask
The uncertainty of volumetric flask is mainly influenced by the three parameter i.e. calibration of the volumetric flask at the time of manufacturing, repeatability and temperature.
Calibration of volumetric flask:
Deviance from nominal volume of 10 mL volumetric flask is ±0.006 mL (at 27°C) as given by manufacturer. Standard value of uncertainty can be calculated with triangular distribution. So, uncertainty related to the liberation of volume by volumetric flask u (V10cal ) is 0.00245.
Repeatability, u (V10rep):
In experiment repeatedly weighing and filling of volumetric flask standard uncertainty established was 0.0014 mL.
The manufacturer has calibrated volumetric flask at time of manufacturing at temperature of 27°C, while temperature at laboratory varied with Δt=±4°C. This difference can be overcome by calculating uncertainty value with estimation of temperature range and volume dilation coefficient. Volume expansion of liquid was taken into consideration as it is quite higher than expansion of volumetric flask. The volume expansion coefficient, λ, of water is 2.1 × 10-4 /°C. Uncertainty for 10 mL volumetric flask ΔV10 was calculated by Equation 6.
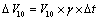 (6)
(6)
Where ΔV10, uncertainty of the 10 mL volumetric flask; V10, volume of the 10 mL volumetric flask; γ, volume dilation coefficient; Δt, temperature variation in the laboratory.Thus, we obtain uncertainty for volumetric flask of 10 mL is 0.0084 mL, standard uncertainty due to temperature on liberation of volumetric flask was found to be 0.0048 mL.
Uncertainty associated with the sample mass msample
Sample mass has three types of uncertainty sources sensitivity, linearity, and repeatability. Mass of the sample was expressed in kg to convince traceability of results.
Sensitivity:
The difference in weighed mass was in very less range and it was measured on the same weighing balance. Thus uncertainty due to sensitivity of balance can be neglected.
Linearity:
A rectangular distribution was assumed to convert contribution of linearity. It was calculated as Equation 7.
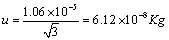 (7)
(7)
Repeatability:
Uncertainty associated with repeatability is found to be 0.0001 kg.
Computation of relative uncertainty due to sample mass:
Using the uncertainty due to linearity and repeatability the uncertainty due to sample mass u (msample) was calculated using Equation 8.
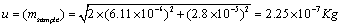 (8)
(8)
Uncertainty associated with concentration, (C10)
Analytical response was collected after each injection in HPLC system of standard solution of different concentration. These responses were used to construct calibration curve. Regression equation of calibration curve was identified such as, slope 9364221442.3334 and intercept 8.8305 for CLZ and slope 13348714077.4857 and intercept 2.4076 for TLM. Uncertainty involved in the construction calibration curve was estimated by injecting 6 different concentration solution each measured three times and sample solution was measured ten times from which Sr and Sxx value were computed as shown in Equation 3 and 4, which were further used to calculate standard relative uncertainty, due to concentration.
For CLZ, Sxx=2.05127 × 10-15
Sr=0.042414
For TLM, Sxx=6.60794 × 10-17
Sr=0.011109
Uncertainty due to recovery of method
Results of recovery are evaluated as percentage recovery from sample matrix after spiking a known amount. When team ‘spike’ is used to estimate recovery, the recovery of analytes from the sample may differ from recovery of spike so that an uncertainty needs to be evaluated. Uncertainty due to spiking is found to be 1.22471 × 10-7 for CLZ and 2.19796 × 10-8. Standard relative uncertainty of method recovery was calculated using uncertainty due to mass of CLZ and TLM (from balance), calibration of pipette, calibration of flask and temperature effect, which was found to be 7.07107 × 10-5, 0.0058, 0.00245 and 0.0048 respectively. Combined uncertainty due to these factor were found to be U (Rf)=0.007917.
Uncertainty due to precision
Method validation results show the repeatability for determination of CLZ and TLM in terms of %RSD (0.08493) and (0.074081) respectively. This equation can be used directly for calculation of CSU.
U (Rep)=RSD
U (Rep) CLZ=0.08493
U (Rep) TLM=0.07408
Combined standard uncertainty (CSU)
The values of all the parameters having effect on CLZ and TLM determination, these are compiled up in Tables 6 and 7 respectively. These values of parameter were further used to calculate CLZ and TLM quantity by using Equation 1 and thus, we obtained a quantity of 4.96 × 10-3 for CLZ and 6.17 × 10-6 mol/kg.
| Formulation | Parameter | Volume, V10(ml) | Sample conc, C10 (M) | Mass samplemsample(kg) | Recovery method | Repeatability |
|---|---|---|---|---|---|---|
| Tablet | Value | 10 | 4.376 ×10-8 | 1.00 ×10-4 | 101 ×10-2 | ---- |
| Standard uncertainity, u(x) | 5.56 ×10-3 | 3.99 ×10-9 | 4.49 ×10-5 | 2.85 ×10-2 | 8.4 ×10-2 | |
| RSU*, u(x)/x | 5.56 ×10-4 | 9.98 ×10-5 | 0.277 | 2.82 ×10-2 | 8.4 ×10-2 |
Table 6: Summary of contribution to the measurement uncertainty for determination of cilostazol through RP-HPLC in tablet dosage form.
| Formulation | Parameter | Volume, V10(ml) | Sample conc, C10 (M) | Mass samplemsample(kg) | Recovery method | Repeatability |
|---|---|---|---|---|---|---|
| Tablet | Value | 10 | 7.83 ×10-9 | 1.00 ×10-4 | 100.7 ×10-2 | ---- |
| Standard uncertainity, u(x) | 5.56 ×10-3 | 1.021 ×10-9 | 2.91 ×10-7 | 2.85 ×10-2 | 7.4 ×10-2 | |
| RSU*, u(x)/x | 5.56 ×10-4 | 1.021 ×10-4 | 0.015 | 2.82 ×10-2 | 7.4 × 10-2 |
Table 7: Summary of contribution to the measurement uncertainty for determination of telmisartan through RP-HPLC in tablet dosage form.
Expanded standard uncertainty (EU)
Expanded uncertainty of CLZ and TLM in sample matrices was obtained by multiplying the combined standard uncertainty by coverage factor k=2 at confidence level of 95%, and, the EU (CLZ/ TLMsample) is as shown.
EU (CLZsample) tab=1.73 × 10-3 mol/kg
EU (TLMsample) tab=7.77 × 10-7 mol/kg
The contribution of different parameter in uncertainty is shown individually for sample matrix has been illustrated in Figure 8.
All analytical endeavors generate measurement data and hence, should necessarily employ appropriate statistical techniques and method of inference, to present and interpret the data. The accurate estimation of variability is challenging. Bayesian approaches offers a different path to the assessment of variability by combining probabilities estimated from detailed study of sub-processes. Developing a new pharmaceutical product requires the designing and testing of manufacturing and measurement process. The resulting process produces quality products when measurements indicative of product quality are on target with minimum variance. In the present study, error propagation break up statistical methods are successfully applied. In this validation was based on the “total error” approach and it can be seen that the method is suited for routine analysis of CLZ and TLM in tablet dosage form with minimum error. In addition, it also illustrates the application of cause-effect analysis in order to estimate the uncertainty in the measuring of CLZ and TLMfrom pharmaceutical formulation through RP-HPLC. The estimation of uncertainty components proved to be a good way for the experimental model to obtain contribution of the uncertainty in the present experiment, concentration of sample is the major contribution towards uncertainty.