Journal of Chromatography & Separation Techniques
Open Access
ISSN: 2157-7064
ISSN: 2157-7064
Research Article - (2017) Volume 0, Issue 0
Horseradish peroxidase (HRP), a versatile heme-containing glycoprotein frequently used in medical diagnostics, is primarily isolated from plant. This process involves filtration, salt precipitation and several chromatography steps, which is expensive, renders low yields and gives isoenzyme mixtures. Although single isoenzymes can be recombinantly produced in Pichia pastoris, they get hyper-glycosylated in the yeast rendering the downstream process cumbersome and thus not competitive. In this study, we analyzed the purification of three HRP isoenzymes differing in the number of N-glycosylation sites recombinantly produced in P. pastoris. We wanted to 1) determine potential correlations between the mode of protein production and the resulting product quality and downstream process, 2) investigate correlations between the number of N-glycosylation sites of HRP and the mode of purification, and 3) find the optimal purification strategy for the recombinantly produced HRP isoenzymes. Thus, we applied different cultivation strategies and tested downstream processes employing both particlebased resins as well as monolithic supports. We showed that the mode of cultivation affected product purity but not the subsequent downstream process. Purification of each of the three HRP isoenzymes using monolithic supports was successful and independent of the number of N-glycosylation sites, whereas purification by particle-based resins was highly affected by glycosylation. Summarizing, we demonstrated a novel feasibility of using monoliths operated in flow through mode for the purification of comparatively small biomolecules.
Keywords: Horseradish peroxidase; Pichia pastoris; Downstream process; Monolith; Particle-based resin
Due to advances in recombinant DNA technology the market for recombinantly produced biopharmaceuticals is booming [1- 3]. Amongst the different recombinant host organisms [4-6], the methylotrophic yeast Pichia pastoris can be used for the recombinant production of complex proteins due to its ability of performing posttranslational modifications [7-11]. However, it is well known that glycoproteins get hyperglycosylated in this yeast [12], which renders the subsequent downstream process difficult and limits the use of the respective product. For some applications, like medical diagnostics, the degree of glycosylation is not as crucial, leaving the problem of purifying hyperglycosylated product from P. pastoris as major bottleneck for the routine use of this organism as production platform.
Horseradish peroxidase (HRP; E.C. 1.11.1.7) is a heme-containing glycoprotein frequently used in medical diagnostics and coupled enzyme assays [13-20]. HRP originates from the horseradish root, where 28 different HRP isoenzymes were identified [21]. Although recombinant production of single isoenzymes in various host organisms has been investigated [22], commercially available HRP is still isolated from its native source. However, extraction from plant is cumbersome, only gives low yields and the final enzyme preparation describes a mixture of isoenzymes rather than single isoenzyme species [23-25] which disagrees with Quality by Design guidelines [26]. Unfortunately, studies on the recombinant production and subsequent purification of this versatile enzyme are scarce. In a recent study, we recombinantly produced 19 individual HRP isoenzymes in Pichia pastoris in shake flasks and developed a 2-step purification comprising a conventional particle-based hydrophobic charge induction chromatography (HCIC) followed by anion exchange chromatography (AEX) using a monolith [27]. We obtained satisfactory results for several recombinantly produced HRP isoenzymes (rHRP) when both purification steps were operated in flowthrough mode. We speculated that hyperglycosylation caused a masking effect of the physico-chemical properties of rHRP and thus prevented interactions with the stationary phases. Less glycosylated contaminant host cell proteins (HCPs) on the other hand interacted with the resins and were retained. Interestingly, we observed a correlation between the number of N-glycosylation sites and the success of flowthrough purification [27]. In fact, these observations motivated us to analyze the purification of recombinant glycoproteins from yeast in greater detail.
Here, we chose three HRP isoenzymes differing in the number of N-glycosylation sites. We recombinantly produced these HRP isoenzymes extracellularly in P. pastoris either in shake flasks using a pulse-wise feeding or in a bioreactor applying a constant feed. Subsequently we compared different protein purification strategies based on either particle-based resins or monolithic supports for purification of the produced isoenzymes. The goals of this study were to 1) determine potential correlations between the mode of protein production and the resulting product quality and downstream process, 2) investigate correlations between the number of N-glycosylation sites and the mode of purification, and 3) find the optimal purification strategy for the rHRP isoenzymes.
HRP isoenzymes and P. pastoris strains
The three HRP isoenzymes were selected based on their differences in the number of N-glycosylation sites (Table 1; [27]).
| HRP isoenzyme | pI | MW[kDa] | N-glycosylation sites | GenBank | UniProt |
|---|---|---|---|---|---|
| A2A | 4.84 | 32.09 | 9 | HE963806.1 | K7ZW28 |
| 1805 | 5.75 | 35.96 | 5 | HE963809.1 | K7ZW05 |
| 5508 | 8.22 | 31.35 | 3 | HE963815.1 | K7ZWW9 |
Table 1: Physico-chemical properties of the three HRP isoenzymes used in this study [27].
All three HRP isoenzymes were recombinantly expressed extracellularly in P. pastoris CBS7435 MutS. The recombinant strains were provided by Prof. Anton Glieder (TU Graz, Austria). A comprehensive description of the identification of the HRP isoenzymes and subsequent generation of the P. pastoris strains has been published before in Ref. [28]. In short, the codon-optimized genes were N-terminally fused to the pre-pro peptide of the alphafactor from Saccharomyces cerevisiae enabling product secretion and the expression of rHRP isoenzymes was regulated by the methanol inducible AOX1 promoter.
Recombinant production of HRP isoenzymes
Recombinant production was done both in shake flasks and in a bioreactor. Three strains, namely rHRP_A2A, rHRP_1805 and rHRP_5508, were cultivated using two different feeding strategies: a dynamic pulse-wise feeding was done during shake flask cultivations, whereas a constant feeding based on the specific methanol uptake rate (qs MeOH; [29]) was applied during bioreactor cultivations (Figure 1).

Figure 1: Schematic overview of recombinant production of individual HRP isoenzymes in shake flasks and bioreactors using different feeding strategies.
Shake flask cultivations: The three rHRP isoenzymes were expressed in 6 × 2.5 L Ultra Yield flasks (BioSilta; Finland). All media were prepared according to the Pichia protocols (Invitrogen; [30]). An overnight culture (ONC) of 50 mL YPD+zeocin in 250 mL Erlenmeyer shake flasks was cultivated at 25°C and 230 rpm for 24 hr. The ONC was transferred to Ultra Yield flasks and a batch phase in 470 mL BMGY+zeocin was done at 30°C and 230 rpm for 24 hr. After that the cofactor hemin was added to a final concentration of 1 mM [31] and an adaptation pulse of 50 mL BMMY was added to each flask. The temperature was reduced to 20°C and 1% (v/v) pure methanol was pulsed every day. Samples were taken every 24 hours to analyze volumetric activity, total protein and RNA concentration in the cellfree cultivation broth. After 120 hours of induction, the cultivation was stopped and the fermentation broth was harvested.
Bioreactor cultivations: Cultivations were done in a 5 L lab scale bioreactor (Infors; Switzerland) and comprised of a batch on glycerol, a fed-batch on glycerol to generate biomass and an induction phase on methanol where the feed was controlled based on qs MeOH [29,32]. To identify qs max MeOH and thus the upper limit of the feeding strategy, dynamic batch cultivations with repeated methanol pulses were performed for each recombinant P. pastoris strain. We have repeatedly described this dynamic strategy to evaluate strain specific parameters before (e.g., [29,33,34]).
Bioreactor cultivations were performed as following: a preculture was grown in YNB+zeocin at 230 rpm and 30°C for 24 hr. Then the preculture was aseptically transferred to 1.5 L sterile 2-fold BSM medium in the bioreactor [30]. The inoculation volume was 10% (v/v). The following process parameters were controlled throughout cultivation: pO2 above 30%, pH at 5.0, temperature at 30°C for batch, fed batch and adaptation phase and at 20°C during induction. Complete consumption of glycerol in the batch phase was indicated by a sharp increase in pO2 accompanied by a drop in CO2 in the offgas signal. A glycerol fed batch was initiated upon batch end with an exponential feeding profile controlled at μ=0.1 h-1. The fed batch phase on glycerol was terminated when the cell density reached 80-90 g·L-1 dry cell weight (DCW). Before adaptation the precursor hemin was added to a final concentration of 1mM [31]. An adaptation pulse of 0.5% (v/v) pure methanol was added to the bioreactor. Upon complete consumption of the pulsed methanol, again monitored in the offgas, the methanol fed batch was started. In analogy to our previous studies [29,35], we performed a fed batch where we stepwise increased the feeding rate corresponding to increasing qs MeOH. The first step was at 25%, the second at 50%, the third at 75% and the last step at 90% of qs max MeOH. Each step was held for 24 hr. This dynamic feeding strategy was shown to result in high productivity before [29].
Processing of cultivation broth
Tthe cultivation broth was centrifuged at 3,500 rpm and 4°C for 30 min. The supernatant containing the rHRP was collected, concentrated 10 to 15-fold and buffer was exchanged for subsequent purification by diafiltration using a 10 kDa cut-off membrane (Omega T-Series; PALL; Austria). Prior to purification, all samples were filtered through a 0.2 μm filter (GE Healthcare; Sweden).
Purification of rHRP isoenzymes
All purifications were conducted on an Äkta Pure25 system (GE Healthcare) at room temperature. We compared two strategies based on either particle-based resins or monolithic supports. For the particle-based strategy we applied MEP HyperCelTM (PALL, Austria) for HCIC followed by a HiLoadTM16/600 SuperdexTM 75 pg column (GE Healthcare) for size exclusion chromatography (SEC). For the monolith-based strategy we used CIMmultusTM C4-HLD for hydrophobic interaction chromatography (HIC) and CIMmultusTM DEAE for AEX, both purchased from BIA separations (Slovenia). For the latter strategy we also investigated the order of the steps. In total, eighteen two-step purification runs were carried out. An overview of the respective experiments is given in Figure 2. For all purification steps the amount of total protein loaded onto the column was kept 20% below the maximum binding capacity of the respective column.

Figure 2: Schematic overview of the experiments conducted in this study.
Particle-based purification strategy: The HCIC column was equilibrated with 20 column volumes (CV) HCIC-A (20 mM sodium acetate, 1 M sodium chloride, pH 8). After 5 CV post-load wash, bound proteins were eluted with HCIC-B (20 mM sodium acetate, pH 8) with a 100% step gradient. The flow velocity during all steps was 100 cm·h-1. The HCIC flowthrough was then concentrated to a volume of 1 mL with centrifugal filters (Ultracel-30 K; Millipore, Ireland) and diafiltrated in SEC buffer (50 mM Bis-Tris, 150 mM NaCl, pH 6.5). The SEC column was equilibrated with 5 CV SEC buffer. The flowrate was 15 cm·h-1. Fractions were collected and pooled by following the absorbance signal at 404 nm indicating the presence of rHRP.
Monolith-based purification strategy: A full factorial multivariate screening with the three factors “type of salt” (NaCl and (NH4)2SO4), “molar strength” (between 1 M and 3 M) and “temperature” (room temperature and 4°C) using the programme MODDE (Umetrics; Sweden) revealed the most suitable buffer compositions. For HIC, the sample was loaded with HIC-A (50 mM Tris-HCl, 2 M NaCl, pH 8) and bound impurities were eluted with HIC-B (50 mM Tris-HCl, pH 8). For AEX, the samples were loaded in AEX-A (50 mM Tris-HCl, pH 8) and bound contaminants were eluted with AEX - B (50 mM Tris-HCl, 2 M NaCl, pH 8). We tested two different ways of using the monolithic supports. The first approach, hereafter called monolith approach A, comprised of AEX as primary purification followed by HIC for polishing. After AEX we simply added salt to the flowthrough and loaded it onto the HIC monolith. In monolith approach B we switched the order of the steps. We used HIC as the first purification step, removed the salt from the flowthrough by diafiltration and loaded the salt-free sample onto the AEX monolith. The flow velocity was 66 cm·h-1 in all these experiments.
Data analysis
Volumetric enzyme activity of rHRP isoenzymes A2A and 5508 was automatically measured by an ABTS assay in a Cubian XC photometric robot (OptoCell, Germany) as described before [36]. Since the maximum reaction rate for HRP isoenzyme 1805 was very low [27], enzyme activity was measured manually using a spectrophotometer (UV-1601; Shimadzu, Long Beach, CA, USA). The manual measurement was carried out at 37°C. 100 μL of 20 mM hydrogen peroxide solution were mixed with 600 μL of 75 mM potassium phosphate buffer, pH 6.5 and 100 μL rHRP. The reaction was started by adding 200 μL of 50 mM ABTS, prepared in 50 mM potassium phosphate buffer (pH 6.5). The change in absorbance was recorded at 420 nm for 600 s. The volumetric activity, aHRP [U·mL-1], was calculated according to equation 1.
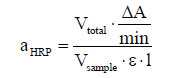 (1)
(1)
Protein concentration was determined by the Bradford assay. The purification factor in the flowthrough (PF) and the recovery of rHRP in percentage (Ra %) were calculated according to equations 2 and 3.
 (2)
(2)
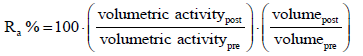 (3)
(3)
RNA concentration was measured using NanoDrop 1000 (Thermo Scientific; USA). Two μL of sample were loaded on the sample pedestal and RNA concentration was measured at 260 nm. Removal of RNA in percentage (RNArem %) was calculated following equation 4.
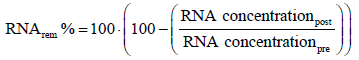 (4)
(4)
All measurements were at least done in duplicates.
Monolithic supports are usually used for the purification of large biomolecules, like viruses, virus like particles [37-39] and monoclonal antibodies [40-43]. Owing to their large channels, convective flow allowing high mass transfer is possible (e.g., [44]). In a previous study we have shown that monoliths operated in flowthrough mode can also be used for polishing of HRP isoenzymes recombinantly produced in the yeast P. pastoris. We found a linear correlation between the number of N-glycosylation sites, which get hyperglycosylated in the yeast, and the success of the flowthrough purification strategy. In the present study we chose three HRP isoenzymes differing in the number of N-glycosylation sites and recombinantly produced them in P. pastoris either in shake flasks or in the bioreactor. We wanted to determine potential correlations between the mode of protein production and the resulting product quality and downstream process, investigate correlations between the number of N-glycosylation sites and the mode of purification, and finally find the optimal purification strategy for the recombinant rHRP isoenzymes.
Recombinant production of HRP isoenzymes
As shown in Figure 1 we cultivated the recombinant P. pastoris strains either in shake flasks or in a bioreactor. During shake flask cultivations we daily pulsed methanol, whereas in the bioreactor we constantly fed methanol following a dynamic feeding strategy [29,34]. For the latter we had to know the maximum specific uptake rate of methanol (qs max MeOH) to avoid overfeeding the cells. Thus, we performed batch cultivations with dynamic methanol pulses [45] to determine strain specific parameters on methanol (Table 2).
| Strain | qs adapt[g·g-1·h-1] | qs max MeOH[g·g-1·h-1] |
|---|---|---|
| rHRP_A2A | 0.006 | 0.042 |
| rHRP_1805 | 0.008 | 0.044 |
| rHRP_5508 | 0.009 | 0.038 |
Table 2: Physiological strain-specific parameters. qs adapt, methanol uptake rate during adaptation, qs max MeOH, maximum methanol uptake rate.
As shown in Table 2, the three recombinant P. pastoris strains had a comparable methanol metabolism, which is why it was possible to apply the same dynamic feeding strategy. In the subsequent fed-batch cultivations we fed corresponding to qs MeOH= 0.01, 0.02, 0.03 and 0.036 g·g-1·h-1 for 24 hours each. In Table 3 we compared the volumetric activity of HRP, the total protein concentration and the total amount of RNA in the cell free cultivation broths at the time of harvest.
| rHRP | Vol. activity [U∙mL-1] | Protein conc. [mg·mL-1] | RNA conc. [mg·mL-1] | Specific activity[U·mg-1] |
|---|---|---|---|---|
| Shake flask | ||||
| A2A | 3.92 | 0.12 | 2.38 | 32.7 |
| 1805 | 0.45 | 0.10 | 2.28 | 4.50 |
| 5508 | 11.5 | 0.10 | 2.29 | 115 |
| Bioreactor | ||||
| A2A | 11.5 | 0.16 | 0.39 | 71.9 |
| 1805 | 0.62 | 0.12 | 0.38 | 5.17 |
| 5508 | 21.5 | 0.12 | 0.33 | 180 |
Table 3: Production of three different rHRP isoenzymes either in shake flasks or in the bioreactor.
As shown in Table 3 the total protein concentrations in cell-free cultivation broths from shake flasks and bioreactor were similar. However, the amount of active rHRP was 2- to 3-fold higher in the bioreactor and the amount of extracellular RNA was around 6-fold lower. Apparently, production in uncontrolled shake flasks applying a pulse-wise feeding is more stressful for the cells resulting in a significant higher amount of contaminants. This is also shown in the specific activity, which can be regarded as a measure of enzyme purity (Table 3).
Purification of rHRP isoenzymes
We tested three different purification strategies using particlebased resins and monoliths and discussed the results in an orthogonal manner, as we compared 1) how the mode of cultivation influenced the DSP, 2) how the number of N-glycosylation sites affected the success of purification, and finally 3) how successful the two different purification strategies were.
Particle-based purification strategy: In a previous study, we developed a simple two-step flowthrough purification strategy for the well-studied isoenzyme rHRP_C1A providing nine N-glycosylation sites. HCIC in flowthrough mode was used for primary purification followed by SEC for polishing [36]. Here, we applied the same strategy for the three rHRP isoenzymes differing in the number of N-glycosylation (N-gly) sites produced either in shake flasks or in the bioreactor (Table 4).
| rHRP | N-glysites | Shake flask | Bioreactor | ||||
|---|---|---|---|---|---|---|---|
| PF | Ra[%] | RNArem[%] | PF | Ra[%] | RNArem[%] | ||
| A2A | 9 | 1.7 | 95 | 91 | 3.0 | 93 | 96 |
| 1805 | 5 | 1.5 | 73 | 98 | 1.0 | 67 | 94 |
| 5508 | 3 | n.a. | 51 | 91 | n.a. | 36 | 86 |
Table 4: Purification of three rHRP isoenzymes produced in either shake flask or bioreactor using particle-based resins.
• Effect of mode of cultivation on DSP
We expected a higher PF for shake flask cultures than for bioreactor broths, owing to uncontrolled conditions, pulse-wise feeding and complex medium and the resulting higher amount of contaminating proteins. However, we did not observe such a trend. In fact, the mode of cultivation did not affect the success of purification, since the amount of rHRP recovered in the flowthrough as well as the amount of removed RNA were comparable independent of the cultivation strategy (Table 4).
• Effect of N-glycosylation sites
As shown in Table 4, the flowthrough purification strategy with particle-based resins worked best for rHRP_A2A providing 9 N-glycosylation sites. There was a clear correlation between the number of N-gylcosylation sites and the amount of product recovered in the flowthrough regardless of the cultivation strategy (Figure 3). The less N-glycosylation sites, the more was the respective rHRP retained on the column, a phenomenon which we had observed before [27]. For rHRP_5508 providing 3 N-glycosylation sites, more than 50% of the product was recovered in the eluate. Therefore, the purification factor in the flowthrough could not be calculated. We concluded that purification of HRP isoenzymes recombinantly produced in P. pastoris using particle based resins in flowthrough mode is only efficient and successful, if the product provides a high number of N-glycosylation sites.

Figure 3: Correlation between the number of N-glycosylation sites and the amount of rHRP recovered in the flowthrough using particle-based resins. Gray squares, rHRP produced in shake flasks; Black circles, rHRP produced in bioreactor.
Monolith-based purification strategy: We used two monoliths, a HIC and an AEX column, for the purification of the three rHRP isoenzymes in this study. We tested two different ways of using the monolithic supports. In monolith approach A we used AEX for primary purification followed by HIC for polishing. After AEX we simply added salt to the flowthrough and loaded it onto the HIC monolith. However, results from initial screening experiments indicated addition of salt to play a dominant role in removal of contaminant proteins. Therefore, in monolith approach B we switched the order of the steps. We used HIC as the first purification step, removed the salt from the flowthrough by diafiltration and loaded the salt-free sample onto the AEX monolith.
Monolith approach A: In Table 5 the purification of the three rHRP isoenzymes using AEX followed by HIC is summarized.
| rHRP | N-glysites | Shake flask | Bioreactor | ||||
|---|---|---|---|---|---|---|---|
| PF | Ra[%] | RNArem[%] | PF | Ra[%] | RNArem[%] | ||
| A2A | 9 | 1.8 | 97 | 84 | 2.7 | 87 | 82 |
| 1805 | 5 | 1.4 | 85 | 99 | 1.7 | 91 | 84 |
| 5508 | 3 | 1.9 | 93 | 76 | 3.4 | 92 | 74 |
Table 5: Purification of three rHRP isoenzymes produced in either shake flask or bioreactor using monolith approach A.
• Effect of mode of cultivation on DSP
Also when using monoliths the mode of cultivation did not seem to affect the success of purification as the amount of rHRP recovered in the flowthrough as well as the amount of removed RNA was comparable independent of the cultivation strategy.
• Effect of N-glycosylation sites
In contrast to particle-based resins (Table 4), all three rHRP isoenzymes could be purified by monoliths independent of the number of their N-glycosylation sites (Table 5). At least 85% of the total amount of rHRP was found in the flowthrough for each of the rHRP isoenzymes. We speculate that the accelerated convective flow through the monoliths [44] impeded product-resin interactions for the less glycosylated rHRP isoenzymes, whereas the slower diffusion in particle-based resins allowed retention.
Monolith approach B: In monolith approach B we changed the order of the monolithic steps and performed HIC followed by AEX (Table 6). In fact, the results were similar to monolith approach A. However, for ease of operation we recommend performing monolith approach A in flowthrough mode to purify rHRP isoenzymes from P. pastoris.
| rHRP | N-glysites | Shake flask | Bioreactor | ||||
|---|---|---|---|---|---|---|---|
| PF | Ra[%] | RNArem[%] | PF | Ra[%] | RNArem[%] | ||
| A2A | 9 | 3.2 | 90 | 94 | 2.8 | 99 | 81 |
| 1805 | 5 | 1.1 | 89 | 92 | 1.5 | 92 | 87 |
| 5508 | 3 | 1.6 | 100 | 84 | 3.2 | 96 | 89 |
Table 6: Purification of three rHRP isoenzymes produced in either shake flask or bioreactor using monolith approach B.
Particle-based vs. Monolith-based purification strategy
The overall PFs for each of the three rHRP isoenzymes were similar when using particle-based resins or monoliths operated in flowthrough mode. However, when particle-based resins were used, rHRP was retained on the column in dependence of the number of N-glycosylation sites (Figure 4).

Figure 4: Correlation between the number of N-glycosylation sites and the amount of rHRP (produced in the bioreactor) recovered in the flowthrough. Gray circles, particle-based purification strategy; Black squares, monolithbased purification strategy.
Summarizing using the monolith-based purification strategy HRP isoenzymes recombinantly produced in the yeast P. pastoris could be purified independently of the number of N-glycosylation sites. We speculate that the accelerated convective flow through the monoliths [44] impeded product-resin interactions for the less glycosylated rHRP isoenzymes. Another huge advantage is the high flow and thus the short process times using monoliths [44]. Using monolith approach A allows an integrated, continuous DSP since the two monoliths can be connected and run in series without a holding step in between, as demonstrated for another product before.
Motivated by previous observations we investigated the use of both particle-based resins and monoliths for the purification of recombinant glycoproteins derived from the yeast P. pastoris. The results of this study can be summarized as:
1) recombinant production in the controlled environment of a bioreactor yielded higher specific activity and lower extracellular RNA concentrations in comparison to shake flasks.
2) the number of N-glycosylation sites played an important role for purification with particle-based resins, whereas it was irrelevant for purification with monolithic supports.
3) with respect to process time, ease of operation and success of purification monoliths outperformed particle-based columns.
In this study we showed that monoliths cannot only be used for the purification of large molecules but also for comparatively small glycoproteins. This purification strategy might describe a platform tool for the purification of recombinant glycoproteins from yeast.
The authors would like to thank BIA separations (Slovenia) for providing the columns and their technical support.
Vignesh Rajamanickam, Maximillian Winkler, Peter Flotz and Lorena Meyer performed experiments. Christoph Herwig and Oliver Spadiut supervised the work. Vignesh Rajamanickam and Oliver Spadiut did the literature search and wrote the article.