Journal of Clinical Trials
Open Access
ISSN: 2167-0870
ISSN: 2167-0870
Review Article - (2012) Volume 2, Issue 2
A critical overview of recent clinical trials in cancer is presently focused on signaling pathway blockers or inhibitors with a view to developing successful clinical trials employing personalized cancer therapies. Rational, pharmacogenomic strategies in cancer trials should be adopted that include specific molecular targeting based on adequate data for, and detailed modeling of, cancer cell genomes, modifications of cancer signaling pathways and epigenetic mechanisms. Novel translational oncogenomics research is rapidly expanding through the application of highly sensitive and specific advanced technology, research findings and computational tools and complex models to both pharmaceutical and clinical problems. Multiple sample analyses from several recent clinical studies have shown that gene expression data for cancer cells can be employed to distinguish between tumor types as well as to predict outcomes. Potentially important applications of such results are individualized human cancer therapies or, in general, ‘personalized medicine’ that will have to be validated through optimally designed clinical trials in cancer. A Human Cancer Genomes and Epigenetics Project is proposed that can provide the essential data required for the optimal design of clinical trials with the goal of achieving significant improvements of the survival rates of cancer patients participating in clinical trials for advanced cancer stages. The results of such a six-year Human Cancer Genomes and Epigenetics Project should also greatly aid with the accelerated, rational development of effective anti-cancer medicines and the chemoprevention of cancers.
Keywords: Human cancer genomes; Human interactomes; Individualized cancer therapy; Differential Gene Expression (DGE); Human lung cancer; Mammalian cell epigenomics; Neoplastic tissue epigenomics; Rational medicine development; Translational oncogenomics; Integrative cancer biology applications in clinical trials; Pharmacogenomics; Tumor cell signal pathways inhibitors; Cancer clinical trials optimization; Cancer patients survival rates; Cancer models; Modular network simulations; Human Cancer Genomes and Epigenetics Project (HCGE)
Recent clinical trials in cancer and organizational trends
The critical evaluation in this review of recent clinical trials in cancer and related methodologies aims at the selection of strategies and methodologies that would optimize the survival rates of cancer patients involved in new clinical trials in the near future. Although very positive and remarkable results were obtained in cancer clinical trials over the last decade, there is ample room for the consideration of various remarkable possibilities and advanced techniques that have only recently become available. Such technological and methodological advances have the potential for substantially improving the survival rates of cancer patients undergoing clinical trials at phase III and IV.
On the one hand, quite remarkable progress has been made with cancer treatments through a handful of such clinical trials over the last decades [1-10], especially with lung cancer treatments where new classes of anti-cancer medicines were thoroughly tested [11], and the development of some of which involved rational pharmacology [12], as in the case of Imatinib for example. Such novel anti-cancer drugs were found to prolong cancer patient lives significantly, and in significant numbers of lung cancer patients. However, many of the drugs tested were shown not to make a significant impact on the growth of malignant tumors, and did not have positive outcomes for cancer treatment. It is therefore surprising that such unsuccessful or unremarkable compounds are currently still being tested in cancer clinical trials in several large countries. Such controversial, cancer clinical trials may be only financially-driven, rather than being rational.
There are already numerous, published reports on several types of clinical trials in cancer and their number is rapidly increasing each year as remarkable progress has already been made over the last decade. However, in this article only a small fraction of clinical trial reports was selected [1-7,13-28] and related research publications [8-12,29-283] because the focus of this article is on optimizing strategies for clinical trials in cancer.
The proposed applications in a significant size project are based on combining advanced methodologies/techniques with conceptual progress via modeling and simulations of complex processes that can lead to neoplastic transformations, malignant tumors and resistance to therapy in clinical trials.
On the one hand, successful complex systems modeling of individual cancers does depend critically upon the availability of sufficiently detailed and reproducible data obtained by such novel techniques and methodologies. The data obtained so far from clinical trials does not satisfy such critical requirements.
On the other hand, successful and effective Pharmacogenomics does require, and relies upon, a sufficiently improved understanding of the molecular mechanisms underlying the development of tumor resistance to treatment in individual cancer patients participating in clinical trials.
The critical need for optimizing clinical trials in cancer
In spite of the remarkable progress made in cancer chemotherapy through clinical trials with novel anti-cancer drugs, the expected ‘magic bullet’ for a complete treatment of cancers has not yet been found, and most of the clinical trials were not optimized for the maximum possible length of survival for the largest number of cancer patients involved in such advanced stage cancer trials. The latter fact raises the important issue of designing rational strategies for clinical trials in cancer that would optimize the survival rates of the maximum possible number of patients undergoing new clinical trials in cancer. The number of new anti-cancer drugs proposed for testing in cancer clinical trials is on the rise, and therefore this issue takes on the urgency of maximizing cancer patients’ survival in such clinical trials. Such an outcome is both highly desirable and also now made possible through multi-disciplinary approaches and high-throughput, low-cost analysis of genomics, interactomics and epigenetics in drug-resistant malignant tumor subpopulations of cancer patients treated in advanced cancer stage clinical trials. The details of such advanced techniques and methodologies, as well as specific recommendations and suggestions, are respectively, provided in Appendix I and II of this article.
The following section 2 presents important concepts related to the control of cell cycling and the development of modular models of cancer interactome networks that serve as a basis for the rational chemotherapy of cancers and for optimizing survival outcomes in new clinical trials in cancer.
Section 3 presents an overview of selected clinical trials in cancer with several signal transduction inhibitors. Additional advanced tools and methodology that are needed for optimizing cancer clinical trials are then presented in Section 3 and Appendix I.
Further required genomic and epigenomic means of optimizing the results and improving the survival outcomes in future cancer clinical trials are, respectively, presented in Sections 4 and 5, and also complemented by Appendix II.
Carcinogenesis is a complex process that involves dynamically inter-connected biomolecules in the intercellular, membrane, cytosolic, nuclear and nucleolar compartments that form numerous inter-related pathways referred to as networks. One such family of pathways contains the cell cyclins. Cyclins are often over-expressed in cancerous cells [101]. This provides a basis for the development of novel rational chemotherapies and chemoprevention of cancers.
A novel theoretical and cancer modeling analysis [58,217] is based on recently published, numerous studies of cyclin signaling, with special emphasis placed on the roles of cyclins D1 and E; our analysis also suggests the possibility of optimizing novel clinical trials through the development of rational therapies of cancer and the possibility of re-establishing cell cycling inhibition in metastatic cancer cells without subsequent transformations that lead to drug resistance.
Cyclins
Cyclins are proteins that link several critical pro-apoptotic and other cell cycling/division components, including the tumor suppressor gene TP53 and its product, the Thomsen- Friedenreich antigen (T antigen), Rb, mdm2, c-Myc, p21, p27, Bax, Bad and Bcl-2, which all play major roles in carcinogenesis of many cancers. Cyclin-dependent kinases (CDK), their respective cyclins, and inhibitors of CDKs (CKIs) were identified as instrumental components of the cell cycle-regulating machinery. CDKs are enzymes that phosphorylate several cellular proteins thus ‘fueling’ the sequential transitions through the cell division cycle. In mammalian cells the complexes of cyclins D1, D2, D3, A and E with CDKs are considered motors that drive cells to enter and pass through the “S” phase. See for example in Figure 1 the gene data related to cyclin D1. Cell cycle regulation is a critical mechanism governing cell division and proliferation, and is finely regulated by the interaction of cyclins with CDKs and CKIs, among other molecules [191].
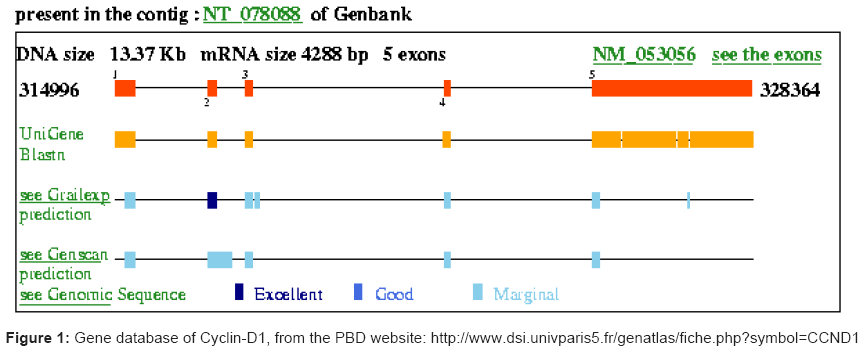
Figure 1: Gene database of Cyclin-D1, from the PBD website: http://www.dsi.univparis5.fr/genatlas/fiche.php?symbol=CCND1
It was also reported that CDKs have another key role –the coordination of cell cycle progression with responses to possible DNA-damage that could, if unchecked or unfixed, lead to a lack of genomic integrity marking the onset of cell disease including cancers [138]. The S-phase is thought to be the most vulnerable interval of the cell cycle because during this interval all of 3 billion DNA bases of the human genome must be replicated precisely in the sense of ‘carbon copies’ being made of the existing DNA strands, without any breaks in the sequence or base substitutions of the copied/replicated strands. Therefore, this correct replication process controls the cell’s survival, especially under genotoxic conditions such as those caused for example by mutagens or X-ray/γ-radiation. Furthermore, Huang et al. reported in [138,139] that CDK mediated the phosphorylation of the FOXO1 transcriptional activator of the proapoptotic genes during the S-phase; when DNA damage occurs either before or during the S-phase, a complex network is activated in the cell which ‘silences’ CDK thereby either delaying or stopping/arresting the cell cycle progression. This may allow the cell to repair the DNA damage by recombination involving BRCA2 and survive. However, if this is not possible because the DNA damage was too great to be reparable, then FOXO1 would trigger apoptosis (cell death). It was proposed that during the unperturbed (normal) S-phase CDK2 phosphorylates FOXO1 at the Serine249 residue in the cell nucleus, which then results in the transfer and sequestering of the FOXO1 in the cytoplasm, where it is well separated from the proapoptotic genes, the ‘target’ of FOXO1 action. Moreover, the CDK-mediated phosphorylation of BRCA2 during the unperturbed S-phase renders inactive the DNA recombination. On the other hand, when DNA becomes damaged, CDK2 is inhibited through the Cdc25A pathway, with the consequence of a dephosphorylated FOXO1 which then remains in the cell nucleus and is able to activate the proapoptotic genes, unless BRCA2 is able to induce DNA recombination and repair in time to prevent apoptosis. The steps that follow are then as explained above: either DNA repair and continued cell cycling, or apoptosis induced by FOXO1. There are still several important questions regarding the entire process that need to be answered before the FOXO1 and CDK2 mechanisms of action can be translated into successful clinical trials based on such knowledge.
A positive correlation has been noticed between overexpression of several cell cycle proteins and unfavorable prognoses and outcomes in several different cancer types [109,128,255]. In human lung tumors and soft tissue sarcomas, it has recently been discovered that cyclin A/cdk2 complex expression and kinase activity were reliable predictors of proliferation and unfavorable prognosis, thereby further substantiating the epidemiological factors of cyclin signaling [94,95,198].
The p21 and p27 proteins
The proteins p27 and p21 are implicated in cyclin regulation and cancer development (Figure 2). Mouse embryonic fibroblasts that were deficient for p27 and p21 were found to contain less cyclin D1 and D2 as well as cyclin D3 [70] than controls. Similarly, mammary glands of p27-deficient mice were shown to possess decreased cyclin D1 levels [5]. It has been demonstrated in vivo that p27 is necessary for maintaining proper levels of cyclins D2 and D3, and this dependency on p27 is common to a wide variety of cells/tissues in vivo. Regarding the molecular interaction between p27 and D-cyclin, CDK4 is a clear candidate as a mediating molecule [74]. Cells employ CDK4/6– cyclin D complexes to flexibly titrate p27 from the complexes containing CDK2, and thereby they control their proliferation. However, mutual dependency between cyclin D and p27 serves also some yet unidentified function in differentiation-related processes. Thus, loss of p27 not only causes unrestricted growth due to inefficient inhibition of CDK2–cyclin E/A, but may also elicit a decrease in levels of D-type cyclins, resulting in differentiation defects. Upon ablation of cyclin D, cells lose their ability to titrate p27 from CDK2–cyclin A/E complexes and proliferation is suppressed. However, defects in differentiation caused by the absence of D-cyclin are reminiscent to defects produced by the absence of p27 [74]. When the changes in levels of p27 and/or D-type cyclins occur, an equilibrium alteration could result between proliferation/differentiation processes that may in the end result in tumorigenesis [74].
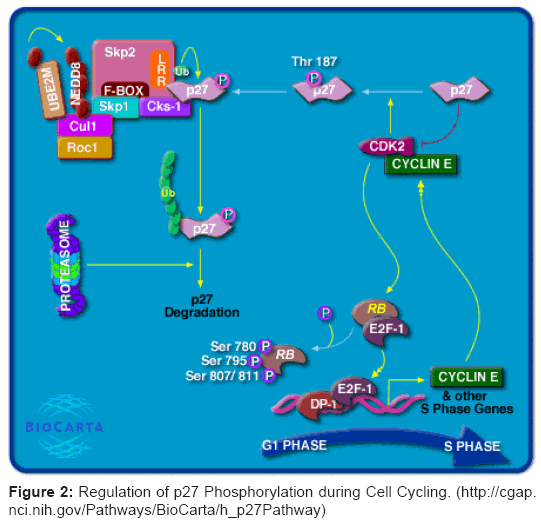
Figure 2: Regulation of p27 Phosphorylation during Cell Cycling. (http://cgap.nci.nih.gov/Pathways/BioCarta/h_p27Pathway)
D1- vs. E- cyclins
The D-type and E-type cyclins control the G1 → S phase transition during normal cell cycling and are important components of steroid- and growth factor-induced mitogenesis in breast epithelial cells [249]. Cyclin D1 null mice are resistant to breast cancer that is induced by the neu and ras oncogenes, which suggests a pivotal role for cyclin D1 in the development of some mammary carcinomas [249]. Cyclin D1 and E1 are usually over-expressed in breast cancer, with some association with adverse outcomes, which is likely due in part to their ability to confer resistance to endocrine therapies. The consequences of cyclin E overexpression in breast cancer are related to cyclin E’s role in cell cycle progression, and that of cyclin D1 may also be a consequence of a role in transcriptional regulation [249]. One critical pathway determining cell cycle transition rates of G1 → S phase is the cyclin/cyclin-dependent kinase (Cdk)/ p16Ink4A/ retinoblastoma protein (pRb) pathway [245]. Alterations of different components of this particular pathway are ubiquitous in human cancer [160]. There appears to be a certain degree of tissue specificity in the genetic abnormalities within the Rb pathway. A model relating Rb to cyclin control in the overall scheme of pro-apoptotic behavior is shown below (Figure 3). In breast cancer these abnormalities include the over-expression of cyclins D1, D3 and E1, the decreased expression of the p27Kip1 CKI and p16Ink4A gene silencing through promoter methylation. These aberrations occur with high frequency in breast cancer, as each abnormality occurs in ~40% of primary tumors. This fact implicates a major role for the loss of function of the Rb pathway in breast cancer. Further details on D1- and E1- cyclins roles in cancer were recently reviewed in [58].
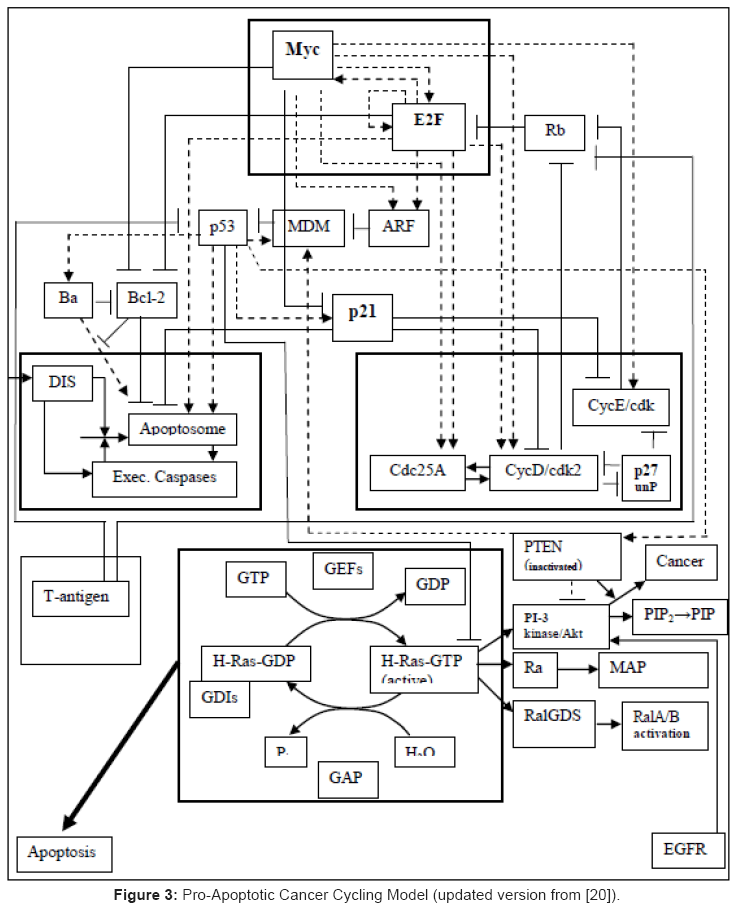
Figure 3: Pro-Apoptotic Cancer Cycling Model (updated version from [20]).
Figure 4: Secondary structures and binding sites of the oligonucleotides HS1 to HS6 and the target RNA.

Figure 5: An illustration of FCCS Applications to DNA Hybridization, PCR and DNA Binding Sites Localization.
Apoptosis and its connection to cell cycle-related proteins is of interest therapeutically, as these types of therapies could ultimately lead to the cancer cell annihilation via apoptosis. Recently, a shift appears to have occurred, with a change in the focus of chemotherapy from exploration of agents that cause cell growth arrest to those that favor apoptosis.
FGFR tyrosine kinases
Fibroblast growth factor receptor (FGFR) tyrosine kinases have recently been studied as they relate to intracellular signaling and their effects on pRb, and are of interest to the field of cancer biology. Overexpression of FGFR tyrosine kinases has been found in many human breast carcinomas and has been associated with poor clinical prognosis [165]. Fibroblast growth factor receptors (FGFRs) are glycoproteins composed of extracellular immunoglobulin (Ig)- like domains, a hydrophobic transmembrane region and a cytoplasmic moiety that contains a tyrosine kinase domain [165]. When active, FGFRs stimulate tyrosine phosphorylation, as well as activation of several signaling molecules: Shc, PI3K, Src, PLCg, Crk, SH2 domain containing phosphatase-2 (SHP-2), p38, STAT1/3 and FGFR substrate 2 (FRS2) [161]. Treatment of tumor cells with the FGFR tyrosine kinase inhibitor leads to a reduction in pRb phosphorylation on serine 795, a site known to be phosphorylated by the cyclin D/cdk4 complex [165]. FGFR signaling may in fact promote cell proliferation by upregulating cyclin D levels. This idea is supported by the fact that ectopic cyclin D1 expression is able to rescue the FGFR inhibitor-mediated antiproliferative effect [165]. Using a cyclin D1 reporter gene, Koziczak et al. found that FGFR inhibitor caused a significant reduction in promoter activity, and was reflected in an overall decrease in cyclin D1 mRNA levels [165].
A recent study employed p27-deficient mice to investigate the significance of p27 for the metabolism of D-type cyclins in differentiated cells [80]. The absence of p27 resulted in decreased cyclins D2 and/or D3 levels in several organs. The drop in cyclin D levels that was due to the absence of p27 equaled the amount of cyclin D physically associated with p27 animal controls. This indicates the possibility that it is the fraction of p27-associated cyclin D that determines the response to p27 deficiency. Cells in which the D-type cyclin level is dependent on p27 do not up-regulate their CDK2 and CDK4 activities upon deactivation of p27 (Figure 2). Moreover, these cells have a negligible amount of p27 bound to CDK2 and/or cyclin A/E under non-cancerous conditions [74]. These findings point to the existence of two roles for p27: regulation of the cell cycle through inhibition of CDKs, and participation in the establishment or maintenance of the differentiated status that is achieved in conjunction with D-cyclins [74], as schematically represented in (Figure 2).
Ubiquitin
The regulation of protein stability via the ubiquitin–proteasome pathway is critical to the comprehension of the biomolecular basis of cancer development. However, ubiquitin modification of substrates signals many cellular processes (besides proteolysis) that are also important for cancer development. Interestingly, many breast cancer proteins studied by clinical researchers are involved in these specific ubiquitin pathways. These proteins include cyclins, CDK inhibitors and the SCF in cell cycle control, the breast and ovarian cancer suppressor BRCA1-BARD1, ErbB2/HER2/Neu and its ubiquitin ligase c-Cbl , as well as and the estrogen receptor and its target, Efp.
One function of the ubiquitin–proteasome proteolysis pathway is to label proteins for rapid degradation. It consists of four enzymes: a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), a ubiquitin ligase (E3) and the 26S proteasome [134]. E1 binds to and activates ubiquitin in an ATP-dependent manner through a thiolester bond and then transfers ubiquitin to an E2 enzyme. E2 then transfers ubiquitin to a lysine residue in the substrate via a terminal isopeptide bond through E3. E3 is a scaffold protein that bridges in the substrate and the ubiquitin-bound E2. The resultant covalent bonds of the ubiquitin ligations form polyubiquitinated conjugates that are quickly found and digested by the 26S proteasome.
A modular, cancer cycling model summarizing various controls involved in the cell cycle is presented in Figure 3.
Understanding these signaling pathways may provide many critical clues toward the development of novel diagnostic tools and treatments for cancer patients [12]. Over the past decade researchers have identified important functional roles for the D- and E- type cyclins in the evolution of human breast cancers. These genes are among the most commonly over-expressed genes in breast cancer, being over-expressed in the early phases of disease and having proven oncogenic effects on mammary epithelial cells both in vitro as well as in vivo. Their established role in CDK activation and Rb pathway regulation has directed scientific attention toward aberrant cell cycling as the basis of oncogenic potential.
More recent data on the role of different G1 cyclins in the areas of differentiation, chromosome stability and transcriptional regulation indicate that their role in breast cancer is much more complex than initially predicted. Further investigations may yield a more complete understanding of the role of these cyclins regarding the biomolecular basis and pathophysiology of breast cancer, with significant potential benefits clinically, through the identification of novel markers of prognosis and therapeutic responsiveness and potential new targets for innovative clinical intervention.
Changes in homeostasis can be followed through various experimental strategies that monitor gene expression profiling, for example, by employing high-throughput microarray technology. This section discusses briefly the successful use of microarray technology in RNA expression studies aimed at identifying signaling pathways that are regulated by key genes implicated in carcinogenesis/ tumorigenesis. A primary objective of tumor-profiling experiments is to identify transcriptional changes that may be the cause of the transition from the normal to the tumor phenotype. Such changes may, however, occur also as a consequence of various neoplastic transformation(s). More importantly, this approach may allow the identification of molecular fingerprints that can be utilized for the classification of different tumor types, and are therefore valuable diagnostic molecular tools in cancer patients. For example, Alizadeh et al. [35] reported that they have successfully used such an approach to identify molecularly distinct subclasses of diffuse large B-cell lymphoma that could not be distinguished by conventional diagnostic tools. In another study, a molecular fingerprint comprising approximately 50 genes has been isolated from a total of over 6,000, and this fingerprint can reliably differentiate between acute myeloid leukemia and acute lymphoblastic leukemia [121]. The approach requires, however, multiple independent experiments with several large groups of samples in order to enable one to reliably and reproducibly separate the biologically relevant changes from false ones that may occur as a result of the genetic heterogeneity between individual samples from the same tumor, for example. The two examples quoted above were able to reproducibly identify tumor type-specific molecular determinants through multiple experiments with various tissue samples.
Identification of specific transcriptional targets in cancer: A different experimental approach to the one presented above is, however, needed for identifying specific targets such as defined genes that are implicated in cancer progression; this involves monitoring changes in transcriptional profile that occur as a result of modulation of the expression level of the defined gene, or genes, selected for such studies. The altered expression profile can be viewed as a ‘blueprint’ by which the defined gene controls its cellular function. The transcriptional profiles are thus employed to define downstream signaling pathways that have been previously validated through other techniques such as differential display [251] and serial analysis of gene expression [280]. This approach combined with microarray technology allows the simultaneous identification of all potential targets. Its only drawback is the reliance upon the prior knowledge of the selected genome for such investigations. The caveat is, however, that the investigator who employs this approach needs also to devise additional experiments in order to confirm that genes identified with the microarray are indeed physiologically relevant targets.
Identification of downstream transcriptional targets of the brca1 tumor-suppressor gene: The breast and ovarian cancer susceptibility gene BRCA1 is probably the most studied gene in the breast cancer field because of its clinical significance and multiple functions. BRCA1 was shown to be mutated in the germline of women with a genetic predisposition to either breast or ovarian cancer [186]. Most mutations identified reported have resulted in the premature truncation of the BRCA1 protein. BRCA1 is known to encode a 1863 amino acid phosphoprotein that is predominantly localized to the nucleus, presumably with a unique function. Protein sequence analysis identified a C-terminal BRCT motif, which was then postulated to play a role in cell cycle checkpoint control in response to DNA damage [162]. Consistent with this postulated role, BRCA1 becomes hyperphosphorylated in response to various agents that damage DNA such as γ/X--ray-irradiation, an effect that was reported to be partially mediated by chk2 kinases [169]. Furthermore, BRCA1 has been shown to be implicated in at least three functional pathways:
i. Mediating the cellular response to DNA damage,
ii. Acting as a cell cycle checkpoint protein, and
iii. Functioning in the regulation of transcription.
However, the physiological significance of such BRCA1 actions as well as their relationships with the function of BRCA1 as a tumor- suppressor gene still remain to be defined. Further details are presented next.
The BRCA1-BARD1 ubiquitin ligase: As already stated above, the BRCA1 gene encodes a 1863-amino-acid protein [186] that consists of a RING-finger domain in its terminal N-region, a region that includes a nuclear localization signal and a domain that binds to many cellular proteins, and tandem BRCT domains in its C-terminal region. BRCA1 is associated with a diverse range of biological processes, such as DNA repair, cell cycle control, transcriptional regulation, apoptosis and centrosome duplication. Thus, a specific role has already been postulated for BRCA1 in transcriptional regulation. The C-terminal domain of BRCA1 was reported to contain a potent transactivation domain when this was fused to a heterologous DNA binding motif [191]. The oligonucleotide array-based expression profiling described above in Section 2.2 was employed in 2000 by Haber in collaboration with Affymetrix Co. to identify the downstream transcriptional targets of the BRCA1 tumor-suppressor gene in order to define its function [130].
The only known biochemical function of BRCA1 is its E3 ubiquitin ligase activity. The N-terminal RING finger domain of BRCA1 interacts with another conformationally similar RING finger protein, BARD1, that also contains an N-terminal RING domain and C-terminal BRCT domains. BRCA1 attains high ubiquitin ligase activity when bound to BARD1 as a heterodimer. Importantly missense mutations in the RING-finger domain of BRCA1 found in familial breast cancer all eradicate the ubiquitin ligase activity of BRCA1-BARD1 [209]. This fact suggests a strong link between BRCA1 ligase activity and its function as a tumor suppressor. The analysis of ubiquitin ligase activity of RING-domain mutations is important not only for the investigation of the biological function of BRCA1, but also to be able to predict a specific patient’s propensity for cancer, which may influence the determination of the need for prophylactic surgery. Besides enhancing BRCA1’s ubiquitin ligase activity, BARD1 is also critical for BRCA1 stability in vivo. Loss of BARD1 leads to a phenotype similar to that of the loss of BRCA1, that is, early embryonic lethality/ chromosomal instability. Moreover, germline mutations of BARD1 are found in both breast and ovarian cancer patients. Although ubiquitin ligase activity may be significant for the role of the BRCA1 gene as a tumor suppressor, the way the activity contributes to BRCA1’s biological function remains unknown. Two issues exist that are critical to the elucidation of the role of the BRCA1-BARD1 ubiquitin ligase: the type of polyubiquitin chain built by BRCA1-BARD1 (and its consequences), and the specific identity of its substrates.
The following reported observations provide only indirect, additional clues to the tumor-suppressor gene function of BRCA1. Germline mutations of BRCA1 were reported for half of breast-ovarian cancer pedigrees and for approximately 10% of women with early onset of breast cancer, uncorrelated with their family history [106]. It was also shown in other studies that somatic inactivation of BRCA1 is rare in sporadic breast cancers [110], and mutations were reported for approximately 10% of sporadic ovarian cancers, therefore suggesting potentially distinct genetic mechanisms for sporadic, breast and ovarian cancers [66]. The reduced BRCA1 protein expression reported for the majority of sporadic breast cancers indicates that epigenetic mechanisms (see also Section 6) may also play a significant role in regulating the BRCA1 expression [268]. Furthermore, a defect was reported in the transcription-coupled repair of oxidative-induced DNA damage in mouse embryo fibroblasts with attenuated BRCA1 function [130]; this observation would suggest that BRCA1 plays a more general role in mediating the cellular response to DNA damage. Thus, BRCA1 has also been reported to be involved in cell cycle checkpoint control, by becoming hyperphosphorylated during late G1 and S cell phases, and then changing to transiently dephosphorylated early after the M- phase [222]. Moreover, the BRCA1 overexpression has been reported to induce a G1/S arrest in human colon cancer cells [9]. By comparison with the cancer regulation model in (Figure 3), it seems very significant for oncogenesis that BRCA1 is physically associated with the transcriptional regulators p53 [202], CtIP [281], c-Myc [264], as well as the histone deacetylases HDAC1 and HDAC2 [277]. The physical association of BRCA1 with c-Myc acquires special significance as c-Myc seems to be involved in controlling telomerase activity, whereas p53 is involved in DNA-repair, cell-cycling and apoptosis. Therefore, in the simplified model presented in Figure 3, one should add the BRCA1 links to both p53 and c-Myc in order to facilitate an understanding of the BRCA1 possible roles in oncogenesis.
Selecting gene expression systems: There are several related problems in studying gene function by expression profiling. For example, it has been often reported to be difficult to generate cell lines that overexpress genes such as BRCA1, or p53, because their forced overexpression can lead either to growth suppression or apoptosis (as shown for example in (Figure 3), and at the end of the previous section). However, in the case of BRCA1, it was reported that the tet-off inducible expression system [124] can be utilized to generate cell lines with highly regulated inducible expression of BRCA1 [131,132]. This inducible expression system introduces into the cells a chimeric transactivator; the latter consists in the tet repressor fused to the VP16 transactivation domain. This chimeric transactivator is inactive in the presence of tetracycline, whereas in the absence of tetracycline it can bind to promoters that contain the tet operator sequence; the latter sequence is then utilized to drive the expression of BRCA1. This expression system has a major advantage in that it allows the change in just one parameter involved in the induction of BRCA1. The BRCA1 induction in one population is the only difference between the genetic backgrounds of the two populations that are being compared by oligonucleotides arrays. A number of BRCA1 transcriptional targets can thus be identified with Affymetrix oligonucleotides arrays, and among these, the stress and DNA damage-inducible gene GADD45 was the gene that exhibited the greatest degree of differential signal intensity [132]. The specific target genes thus identified were also verified by Northern blot or quantitative reverse transcriptase-PCR analysis in order to confirm induction in response to the stimulus, that is, the induction of BRCA1 [132].
Further details were discussed in a recent, related report [58].
In another recent report [278], Yu et al. utilized a modified version of the tet-off inducible expression system to define the downstream transcriptional targets of the p53 tumor suppressor gene [280]. A total of 34 genes were identified that exhibited at least a 10-fold upregulation in response to the inducible expression of p53. Somewhat surprisingly, there was a marked heterogeneity of the response when it was evaluated in different cell lines derived from the same tissue of origin. Among the 33 genes studied only nine were found to be induced in a panel of five unrelated colorectal cell lines, and 17 were induced in a subset; however, eight were not induced at all in any of the five cell lines examined. This can be interpreted as being due to a high degree of cell type specificity. Furthermore, p53 was not absolutely required for induction for the majority of the genes identified in response to either adriamycin or 5-FU. Therefore, these agents do not seem to act exclusively through p53, suggesting that there is inherent redundancy in the majority of signaling pathways. Such inherent redundancy in signaling pathways of cancer, and untransformed, cells might be important in understanding the results of clinical trials in cancer treatment with signal transduction modulators that will be discussed in the next (subsection (3.2)).
Clinical trials with signal transduction inhibitors - novel anticancer drugs active in chemo-resistant tumors
Recently, there is an increasing number of reports suggesting that human cancers frequently involve pathogenic mechanisms which give rise to numerous alterations in signal transduction pathways. Therefore, novel therapeutic agents that target specific signal transduction molecules or signaling pathways altered in cancer are currently undergoing clinical trials often with remarkable results in cancer treatments of patients in which chemo- and/or radio- therapy resistant tumors have become apparent. For example, several classes of such anti-cancer drugs that were developed during the last decade are:
• Tyrosine/threonine kinase inhibitors, including: STI-571 (‘Gleevec’, or Imatinib Mesylate), ZD-1839 (‘Iressa’), OSI-774, and flavopiridol, which are ATP-site antagonists and have already completed phase I and phase II trials; Imatinib Mesylate and Iressa are FDA approved.
• Several other kinase antagonists that are currently undergoing clinical evaluations, including UCN-01 and PD184352;
• Other strategies for downmodulating kinase-driven signaling include 17-allyl-amino-17 demethoxygeldanamycin and rapamycin derivatives. Phospholipase-directed signaling may also be modulated by alkylphospholipids.
• Farnesyltransferase inhibitors, originally developed as inhibitors of ras-driven signals, may attain anti-cancer activity by affecting other/or additional targets; several are FDA approved;
• Monoclonal antibodies (Mab-class) Herceptin and C225; FDA approved.
Medicines that block in a highly specific manner the signal transduction in cancer cells are an efficient method for fine-tuning the development of effective cancer treatments if the development of resistance to such drugs through mutations can be either prevented or circumvented by second and third generation medicines whose action is not affected by the multitude of possible mutations in a malignant tumor. The following detailed background on clinical trial and signal transduction modulators as novel anticancer drugs summarizes the contents of an earlier NCI Report of such signal transduction inhibitors in cancer cells [20].
Tyrosine kinase inhibitors: T1. STI-571, or ‘Gleevec’, or Imatinib mesylate:
I. STI-571 action mode and impact:
a. Inhibits three kinases: Abl (all forms), PDGFR and c-kit tyrosine kinases;
b. Blocks the Bcr-Abl tyrosine kinase;
c. important in chronic myelogenous leukemia (CML) patients because CML cells have constitutively active Bcr-Abl tyrosine kinase;
d. STI-571 differentially inhibited the growth of p210Bcr-Abl CML and p185Bcr-Abl CML containing acute lymphoblastic leukemia cells and does not affect the normal marrow cells;
e. The effect of STI-571 is truly exciting because it inhibits c-kit/CD117 positive tumors where there is a paucity of interventions for such chemoresistant tumors. An example of its action: a significant response was observed in rapidly progressive gastrointestinal tumors (GIST) and also in soft-tissue sarcomas that were previously resistant to several cytostatic, anticancer drugs when Gleevec was not administered simultaneously with such cytostatics;
f. FDA has approved Gleevec for GIST as well as CML treatments, and is undergoing clinical trials for novel therapeutic strategies of other types of cancer.
g. This is a remarkable success example of clinical trials for cancer treatment.
II. T2. SU5416:
h. This ATP-site antagonist of the vascular endothelial growth factor (VEGF) (Flk1/KDR) receptor was designed following studies of the indolin-2-one pharmacophore and the fibroblast growth factor (FGF) receptor tyrosine kinase domain. A Lineweaver-Burk analysis showed SU5416 to be a competitive inhibitor with ATP for the Flk1/KDR and PDGF receptors (Ki ~0.16 μM and 0.32 μM, respectively), as reported in [16] and [189].
i. The first SU5416 clinical trial enrolled 63 patients and administered the drug i.v. biweekly [222]; at the higher doses, nausea, vomiting, headache and some liver toxicity were noticed; [stable disease of greater than 6 months duration was the only reportable outcome in patients with a variety of advanced diseases (colorectal, lung, renal and Kaposi’s sarcoma);
j. Patients with significant progression suffered noticeable increases in vascularity; the occurrence of vascular complications like thrombotic events raises the risk of broad application of this drug [167].
III. Tyrosine kinase/EGFR inhibitors:
a. 5.2.2.1) TE1. ZD 1839 (‘Iressa’) action mode: EGFR, the Epidermal Growth Factor Receptor (s) activate(s) several downstream signaling pathways and is over-expressed in numerous types of human cancers, including: non-small cell lung (NSCLC), colorectal, head and neck, bladder, brain, pancreas, breast, ovary, prostate, and gastric cancers [126,225]. Overexpression of EGFR is associated with increased invasiveness, resistance to treatment and poor outcomes in several tumor types [157,197] specified herFound to be effective in the treatment of: Non-small cell lung (NSCLC), colorectal, head and neck, bladder, brain, pancreas, breast, ovarian, prostate and gastric cancer types that were previously unresponsive to other chemotherapy [126,226];
b. ZD 1839 (Iressa) blocks EGFR; ZD1839 inhibits autophosphorylation, and resulted in complete regression in some xenograft tumors [82,245] when used either in conjunction with cytotoxic drugs such as doxorubicin, or in combination with radiation; Iressa inhibits the Ras/MAP kinase and STAT-3 transcription factors, in many tumors; the inhibition of the epidermal growth factor receptor (EGFR) has been of significant interest lately, partially because of the autocrine activation of EGFR and several downstream pathways, such as the ras/MAP kinase and STAT-3 transcription factors, in several tumors. The activated EGFR pathway induces entry into the cell cycle, inhibition of apoptosis, and also activation of angiogenesis and motility. Several phase I and II studies with Iressa have already been completed [2,63,198]. Daily oral doses have ranged from 50 to 700 mg for 2 to 4 weeks. ZD1839 resulted in some responses in NSCLC and prostate cancer, and stability of disease (over 4 months) in several patients [63,111,198]. 22% of Japanese patients achieved partial response according to Negoro et al. report in [198]; side effects have been relatively mild and have included diarrhea and rash.
IV. TE2. OSI-774 ( Erlotinib, or ‘Tarceva’): ‘Tarceva’ is also an EGFR inhibitor; it binds very tightly to EGFR, causing EGFR inhibition, and also produces downstream inhibition of the P13/MAPK signal transduction pathways, resulting in accumulation of p27, that leads to cell cycle arrest in the G1 phase, and thus causes induction of apoptosis [196]. EGFR-TK is more than 1000- fold sensitive to ‘Tarceva’ compared with any other tyrosine kinases. Therefore, it is a very specific inhibitor of EGFR –TK and reduces very markedly the phosphorylated EGFR-TK;
a. The IC50 for ‘Tarceva’ is 2 nM (when measured by purified EGFR-TK inhibition in biochemical assays), and its value is 20 nM for the EGFR-TK autophosphorylation when measured in intact cells;
b. Proposed mechanism of action: reversible inhibition of EGFR-TK through competitive binding to the ATP site;
c. Results of preliminary clinical trials reports are: partial responses in patients with colorectal cancer and renal cell carcinoma (kidney), as well as > 5 month stabilization in: colon, prostate, cervical, NSCLC and head and neck cancers.
V. TE3. ‘Herceptin’ (or Trastuzumab) action modes and impact:
a. Trastuzumab, a recombinant humanized monoclonal antibody directed against HER2, is known as ‘Herceptin’ [51]. The HER2/neu gene increases the kinase activity, initiating signal transduction, that leads to proliferation and differentiation in approximately 30% of human breast cancers (with as many as 50 to 100 gene copies/cell);v
b. The HER2/neu gene makes a type I receptor tyrosine kinase encoding a 185 kDa surface membrane receptor protein;
c. Phase I trials showed that the dose of trastuzumab (i.v. at 10 to 500 mg single dose or weekly) could be increased without toxicity and that pharmacokinetics were dose-dependent [239]. Phase II trials response was >5.3 months. Phase III trial patients received doxorubicin or epirubicin plus cyclophosphamide; 28% of patients treated with chemotherapy and trastuzumab were reported to be free of tumor progression, compared with 9% of the patients treated with standard chemotherapy alone.
d. The monoclonal antibody of the membrane receptor HER2 signaling protein was reported to be much more efficient than chemotherapy alone. About 1 in 5 of the patients had cardiac dysfunction, when trastuzumab was administered at 4 mg/kg body weight initially; this is, therefore, a clear example of a clinical trial that was not optimized for patient survival rates even though at phase II 28% of the patients treated were cancer progression-free one year later.
e. A phase II trial was conducted with 46 HER2 (+) metastatic breast cancer patients who had failed prior cytotoxic chemotherapy [35]. Objective responses were seen in 5 of 43 assessable patients, including one complete remission and 4 partial remissions. A second phase II trial combined trastuzumab with cisplatin in 39 HER2 (+) metastatic patients who had failed prior chemotherapy [211]. Of the group of 37 subjects, 9 achieved a partial response and 9 had a minor response or stability. A randomized, placebo-controlled phase III study was performed to determine efficacy and safety of adding trastuzumab to chemotherapy in breast carcinoma. 28% of patients treated with both were disease progression-free at 12 months, compared with 9% of the patients treated only with ‘standard’ chemotherapy.
f. The treatment is indicated as a single agent for patients that have failed earlier therapy, and it is used also as first-line treatment for metastatic disease when applied in combination with paclitaxel.
g. Trustuzumab has already been approved by the FDA for use in women with metastatic breast cancer with HER2-positive tumors.
VI. TE4. Cetuximab: An antibody-based approach to affecting tyrosine kinase signaling is by cetuximab, a humanized monoclonal antibody against the EGFR. Mab225, a murine monoclonal antibody that specifically binds to EGFR, specifically competes with signal transduction initiated by TGF-α [116]. Cetuximab (C225) is a human-mouse chimeric version of Mab225 that binds specifically to EGFR with high affinity, thus preventing the ligand from interacting with the receptor. Preclinical studies show that cetuximab results in cell-cycle arrest, as well as apoptosis, in different contexts [140]. A synergistic effect of cetuximab with cytotoxic chemotherapy has been seen with cisplatin, doxorubicin [63], gemcitabine [73], docetaxel [254], and paclitaxel [144]. Early phase I trials demonstrated that cetuximab displays nonlinear, dose-dependent pharmacokinetics that are not altered by coadministration of cisplatin [62]. These studies were conducted in patients with tumors overexpressing EGFR. There were only 5 episodes of severe C225-related toxicities among the 52 patients. Two patients with head and neck tumors who received cetuximab at doses of 200 mg/m2 and 400 mg/m2 with cisplatin exhibited a partial response. In light of these results, the clinical development of cetuximab is continuing with a number of phase II and III studies.
Serine-threonine kinase antagonists (Stkas)
S1. Rapamycin congeners:
a. Rapamycin (Sirolimus, Rapamune) is a macrolide fungicide that binds intracellularly to the immunophilin FKBP12; the resulting complex inhibits the activity of a 290-kDa kinase known as mTOR – the mammalian target of rapamycin. Rapamycin was isolated from the bacterium Streptomyces hygroscopicus and possesses potent antimicrobial and immunosuppressive properties.
b. Sirolimus was approved by the FDA for the prevention of allograft rejection following organ transplantation in humans.
c. Several subsequent studies with rapamycin revealed significant antitumor activity. This is as expected because of the importance of mTOR in mitogenic cell signaling. The mTOR is a kinase member of the PI3K-related kinase family which is activated in response to growth signaling through the PI3K/Akt pathway. The activation of mTOR results in an increased translation of several critical cell-cycle regulatory mRNAs through two downstream effector kinases, p70S6K and 4E-BP1/PHAS [118,240]. Rapamycin causes G1 cell-cycle arrest by increasing the turnover of cyclin D1 [134], preventing upregulation of cyclins D3 and E [88], upregulating p27KIP1, and also inhibiting cyclin A-dependent kinase activity [156]. Several analogs of rapamycin have been selected for further development as anticancer agents.
d. CCI-779, an ester of rapamycin, has significant antiproliferative effect and favorable toxicology profile and is being studied in several phase I trials in humans [137,218]. Several partial responses have been documented in renal cell carcinoma, NSCLC, neuroendocrine tumors, and breast cancer, in addition to minor responses or stable disease in several tumor types [137,218]. RAD001, an orally bioavailable hydroxyethyl ether derivative of rapamycin, also has potent activity against various animal xenograft models of human tumors; an antiangiogenic effect may account in part for its antiproliferative properties [203].
S2. MEK inhibitor PD 184352:
a. The stimulation of Ras-mediated signal pathways results in a cascade of downstream kinase activation including Raf, which phosphorylates two distinct serine residues on the dual-specificity kinase MEK (MAP kinase-kinase) [151]. MEK, in turn, activates and exclusively phosphorylates two subsequent kinases, ERK1 and ERK2 (MAPK), on specific tyrosine and threonine residues within each kinase. These kinases phosphorylate a variety of substrates including transcription factors critical to cell proliferation and tumor invasion [161].
b. In cytotoxicity studies, correlation between sensitivity to PD184352 and increased activated MAPK levels was observed in some cells-in particular, colon cancer cells. Higher levels of MAPK activation were observed in colon tumor tissue versus normal mucosa as this event occurs late in colon carcinogenesis [236].
c. In mice with colon 26 xenograft model treated with PD184352, excision and assay of tumor cells revealed diminished phospho-MAPK levels. After drug withdrawal, a return to baseline levels was observed reflecting the cytostatic nature of the inhibition. The pharmacodynamic measurement of activated MAPK in tumor tissue may be used as a biological marker of drug activity as antibodies specific for phosphorylated MAPK are routinely available.
S3. Bryostatins:
a. The bryostatins represent a large family of secondary metabolites produced in extremely small amounts by the marine invertebrate, Bugula neritina of the phylum Ectoprocta [215]. The various bryostatins are distinguished by varying side chains off the macrocyclic lactone ring structure. Despite this close structural relationship, these nontumor-promoting PKC activators have different biologic activities and spectrum of toxicity [149,166]. Bryostatin 1 (Bryo1) is the prototype of this 17-member family and the most extensively studied in humans. Initial isolation of Bryo1 was based on its antineoplastic activity against the murine P388 lymphocytic leukemia. Bryo1 is a potent and rapid activator of PKC; however, unlike other PKC activators, including phorbol myristate acetate (PMA), Bryo1 lacks tumor-promoting capabilities.
b. The first two published phase I trials evaluated Bryo1 administered as a 1 h intravenous infusion [211,216]. The DLT was myalgia, occurring approximately 48 h after treatment and lasting up to several weeks at the highest dose levels (65 μg/m2/dose). The MTD was 50 μg/m2, and the recommended dose for phase II trials was 35 to 50 μg/m2 every two weeks. Partial responses were observed in two patients with malignant melanoma, which lasted 6 months and 10 months. Plasma levels of tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6) increased 2 h and 24 h after treatment, respectively, and were dose related.
S4. UCN-01 (7-OH staurosporine):
a. Staurosporine, a natural product isolated from Streptomyces staurosporeus, is a relatively broad action, nonspecific protein kinase antagonist, originally isolated in an effort to define inhibitors of protein kinase C (PKC). 7-OH staurosporine (UCN-01) was defined as a more selective, but not specific, PKC antagonist.
b. Two prominent effects of UCN-01 have emerged in preclinical studies in vitro: induction of cell-cycle arrest, and abrogation of the checkpoint to cell-cycle progression induced by DNA damaging agents. UCN-01 inhibited cell growth in several in vitro and in vivo human tumor preclinical models [32]; however, antiproliferative activity on the part of UCN-01 cannot be explained solely by inhibition of PKC. Firstly, in cell-cycle analyses UCN-01 inhibits Rb+ cells at G1/S phase of the cell cycle [33]. Secondly, cells treated with various concentrations of UCN-01 showed decreased pRb phosphorylation in a dose-dependent manner [81]. These results suggest that CDK2- or CDK4-regulated steps are targets for UCN-01-induced cell-cycle arrest.
c. UCN-01 abrogates the DNA damage-induced checkpoints to cell-cycle progression in G2 [76,261], and in S phase [240]. It is noteworthy that these effects were apparent at drug concentrations that appeared to have little direct effect on cell proliferation or that caused enhanced cytotoxicity by clonogenic or proliferation assays. In addition, they provided a mechanistic framework for prior observations that DNA-damaging agents such as mitomycin [32] could greatly potentiate UCN-01 action.
d. In contrast to animal studies, UCN-01 displayed strong binding to human plasma proteins, apparently to the α1-acid glycoprotein (AAG) in initial human phase I clinical trials [20,112]. One partial response occurred in a patient with melanoma, and a protracted (>4 year) period of stabilization of minimal residual disease was observed in a patient with alk (+) anaplastic large cell lymphoma.
Miltefosine and perifosine (ALP analogs):
a. Certain alkylphospholipids (ALP) (e.g., Rac-1-O-octadecyl-2-O-methyl-glycero-3-phosphocholine [ET-18-OCH3, edelfosine]) when given to mice prior to transplantation of Ehrlich ascites carcinoma cells, effectively prevent growth of this tumor [252]. Enhancement of immune defense against tumor cells was initially considered a plausible mechanism and has been demonstrated on multiple occasions by a number of ALP analogs.
b. Edelfosine is also able to induce apoptosis in HL60 leukemic cells, even in low concentrations and after short incubation times. In U937 leukemic cells, the compound induced apoptosis rapidly, whereas in epithelial HeLa tumor cells this induction required prolonged times of treatment [191].
c. All ALP analogs studied so far cause an indirect inhibition of PKC, most likely as a result of the reduced formation of diacylglycerol through inhibition of phospholipase C [237,255]. Additional antiproliferative mechanisms could involve altered growth factor receptor function, as well as recent evidence of p21 induction by an as yet undefined pathway [208], irrespective of p53 function.
d. Eight phase I-II studies, consisting of 443 patients using topically applied miltefosine 2%-8% for skin metastases in patients with breast cancer, showed a median response rate of 38% [24,26,27]. Evidence from the trials led to the approval of miltefosine, licensed as Miltex©, in Germany for the treatment of cutaneous breast cancer and cutaneous lymphomas.
e. The heterocyclic alkylphosphocholine derivative octadecyl-(1,1-dimethyl-piperidino-4-yl) phosphate (D-21266; perifosine) was developed and selected for improved gastrointestinal tolerability. A number of phase I studies with this compound have been completed both in Europe and USA; early evidence points to greater tolerability and less gastrointestinal toxicity [186].
The proteasome inhibitor PS-341:
a. The proteasome, a multicatalytic protease responsible for degradation of most proteins with the cell, has emerged as a new target for anticancer drug development. The 20S proteasome is involved in the degradation of several cell-cycle regulatory proteins such as cyclins (A, B, D, E), cyclin-dependent kinase inhibitors (p21WAF1/CIP1 and p27), oncogenes (c-fos/c-jun, c-myc, N-myc), p53 and regulatory proteins (IκB, p130) [159]. Inhibition of the 20S proteasome pathway, therefore, aims at altering the cell cycle to promote apoptosis [39]. Although the proteasome is present in all cells, transformed and dividing cells are most sensitive to its inhibition [96].
b. PS-341 is the first proteasome inhibitor to enter human trials. It is a boronic acid dipeptide that specifically inhibits the 20S proteasome presumably through the stability of a boron-threonine bond that forms at the active site of the proteasome. It was found to have substantial cytotoxicity against a wide range of human tumor cells in the NCI 60 cell line anticancer drug screen [29]. PS-341 causes accumulation of cyclin A, cyclin B, p21WAF1/CIP1, and wild-type p53 and arrests the cells at the S and G2/M phases followed by nuclear fragmentation and apoptosis. PS-341 significantly inhibited NF-B DNA binding and functional reporter activity [250]. Several phase I studies evaluated various schedules of PS-341 administration. At the MTD recommended for phase II studies (1.25 mg/m2-1.3 mg/m2), a 65%-72% inhibition of 20S proteasome was achieved [13,30]. An average 54% inhibition of proteasome was achieved in patients’ tumors [127]. In these phase I studies several patients achieved partial responses and disease stabilization including a bronchoalveolar NSCLC, melanoma, sarcoma, lung adenocarcinoma, and malignant fibrous histiocytoma. Patients usually had more toxicity with the second cycle of treatment. Currently several phase II clinical trials are evaluating PS-341 as a single agent in hematologic malignancies, neuroendocrine, renal cell, melanoma, breast, brain, pediatric tumors, and several other solid tumors. Significant antitumor effects were documented in a phase II study of PS-341 in refractory multiple myeloma [219].
Farnesyl transferase inhibitors:
a. Ras genes are mutated in 30% of all human cancers with K-Ras being the most common. This family of genes encodes GTP binding proteins that are important in malignant transformation, cell growth, and intracellular signal transduction.
b. Normal ras binds GTP and in the GTP-bound state interacts with numerous effectors including the raf proto-oncogene kinase and phosphatidyl-inositol 3-kinase.
c. Three isoforms, Harvey (Ha), Kirsten(K), and N-isoforms have been described, with mutation of the GTPase of the K isoform resulting in a persisting signaling capacity in approximately 20% of human epithelial tumors. N-ras is mutated in a smaller proportion of malignancies, predominantly leukemias. Ras function requires lipophilic anchorage to the cell membrane by lipid prenylation. This requires posttranslational modification or covalent thioether bond formation between a farnesyl group (C15) and a cysteine residue at the ras carboxy terminus. A “GTT shunt pathway” maintains K-Ras in an active prenylated, membrane-bound form and explains in part the requirements for higher farnasyl transferase inhibitor (FTI) dose or co-treatment with a GTT inhibitor for significant growth inhibition in K-Ras models [210]. Several classes of FTIs have been developed in an initial effort to define inhibitors of Ras function and, in general, compete with the enzyme substrates, the CAAX tetrapeptide, and farnesyl pyrophosphate (FFP). The CAAX competitors are generally peptidomimetic agents that mimic the carboxyl terminal portion of the Ras protein.
F1. SCH66336:
a. SCH66336 is a novel oral agent derived from a class of nonpeptide, nonthiol-containing, CAAX mimetic FTIs [67]. The drug inhibits in vitro FT activity with an IC50 of 1.9 nM for H-ras, 2.8 nM for N-ras, and 5.2 nM for K-ras. Inhibition of cells with activated ras and anchorage-independent growth was noted with IC50 of 75 nM in H-ras versus 400 nM with K-ras-driven cells [176]. The observed growth inhibition of tumor cells in soft agar and in xenografts was independent of ras mutational status because even wild-type ras cells were sensitive [177].
The phase I experience with SCH66336 involved 20 patients using a twice a day schedule over 7 days every 21 days. Eight patients had stable disease, and treatment for up to 10 cycles was possible in a few patients. Antitumor activity was reported in only one patient with advanced NSCLC who had a greater than 50% reduction in an adrenal metastasis and received treatment for 14 months [1]; further studies will be needed as statistical significance had not been achieved in this clinical trial.
F2. R115777:
R115777 is a substituted quinolone and competitive inhibitor of the CAAX peptide binding site of FT [103]. The compound inhibits in vitro K-Ras farnesylation (IC50 7.9 nM) and exerts antiproliferative effects in cell lines such as H-Ras-transformed fibroblasts (IC50 1.7 nM) and K-Ras-driven colon and pancreatic cells lines (at roughly IC50 20 nM) [104].
a. The initial clinical experience with R115777 in 27 patients was reported by Zujewski et al. [28]. A patient with metastatic colon cancer had symptomatic improvement and a 50% reduction in carcinoembryonic antigen (CEA) levels.
b. A most interesting outcome was obtained in patients with myelo-dysplastic syndrome or relapsed or poor prognosis leukemias, where a phase I dose escalation study revealed DLT at 1200 mg twice per day, consisting of neurotoxicity, with non-DLTs including renal insufficiency and myelo-suppression. There was clear evidence of down-modulation of erk kinase activity, along with the farnesylation status of lamin A and HDJ-2. Clinical responses occurred in 29% of 34 evaluable patients, including 2 complete responses [154]. Though there were no mutations in N-Ras detected in this patient population, this study did suggest that in addition to clinical activity there was some evidence of down-modulation of signaling as well as farnesylation-directed activities.
The results summarized in this section convey the promise, as well as some of the major challenges, still encountered in developing effective signal transduction inhibitors for cancer treatment. These molecules represent a distinct departure from previous therapeutic approaches based on cytotoxic activity in tumor models, without reference to underlying mechanism. The fact that any responses have been seen at all reaffirms the relevance of tumor cell biology in charting the further course of cancer developmental therapeutics. However, the initial experiences raise a number of issues that should be considered before the field advances.
First of all, with certain agents the actual magnitude of conventionally described responses is lower than would usually be associated with clinical value. A more accurate means of diagnosing the dependence of a tumor on a particular signaling pathway or target must be defined. Microarray, proteome- and interactome- based approaches offer the promise of personalized cancer therapies, but these must be integrated into the clinical trials process in a manner that optimizes the survival rate of the cancer patients. Some agents have entered initial clinical trials with extensive efforts to document target-based effects in conjunction with pharmacology and clinical toxicity evaluations. On the other hand, other agents have not, however, been sufficiently evaluated before beginning the clinical trials, and in those instances the phase I study may lack depth as far as patient survival and also deriving valuable therapeutic information is concerned. Lacking clear evidence of clinical response, one cannot recommend in such cases to move forward to the next phase.
Intelligent design of combinations with standard cytotoxic agents also remains a significant challenge because the cytotoxic treatments are well-established in medical practice. Pre-clinical models of synergistic effect with signaling agents often proceed from empirical considerations without a rational basis that would guide clinical implementation, for example, by employing in silico simulations with modular models (Figure 3) that can predict the qualitative dynamics of cancer cell lines in a tumor when sufficient genomic information is available from microarray determinations. Such circumstances therefore call for renewed efforts to define better assays of target effects in the pre-clinical phase of a drug’s development that can be rationally translated to the clinical arena.
Many of the agents utilized both in model systems and in initial clinical observations on cancer patients might be associated with protracted periods of disease stability, rather than overt cytotoxic effects which might be assigned to the initiation of an apoptotic response. Though such stable disease cases can be readily observed in animal tumor models, it is uncertain whether these could be meaningfully captured also in clinical populations of human patients with advanced forms of cancers, and especially with treatment-resistant tumors. One must develop decision-making, step-by-step strategies that will also aid in the use of potent drugs in patients at the earlier cancer stages, or indeed design prevention and adjuvant strategies. Clinical study algorithms for various cancers, and especially lung cancers, must also be developed that address such biologically relevant possibilities in a manner that does not compromise patient safety, and at the same time maximizes the rate of patients survival in cancer clinical trials.
There is clearly a need for individualized cancer therapy strategies based on high throughput microarray information recorded for isolated tumor cell lines from stage I through stage III cancer patients; such data is essential for improving the survival rate of stage III cancer patients undergoing clinical trials with novel signaling pathway modulator or blocker medicines, such as those discussed in this section. Specific methodologies, advanced techniques (see Appendix I) and suggestions for developing such strategies for personalized cancer treatments are discussed in the next sections and the two appendices.
It has been claimed that high-throughput yeast-two-hybrid (HT-Y2H) methods will allow a systematic approach to functional genomics, by placing individual genes in the global context of cellular functions [164]. One finds that high-throughput screening methods such as HT-Y2H have indeed allowed the mapping of the first interactomes for three eukaryotes [119,174]. Because of the human interactome’s much larger size and its very high-degree of complexity there will be quite high costs and labor involved in obtaining the data necessary, for example, for an HT-Y2H mapping of a complete human cell interactome. Furthermore, the complete data analysis together with the assembly of the complete interactome network is likely to require both conceptual and computational advances, in addition to a significant amount of time and collective effort(s) by one or several research teams. In view of the high, potential importance of the human interactome for cancer therapy, and also for improved diagnosis and ‘rational’ clinical trials, such an effort should now be the top priority or, at the very least, must be given a priority far above that of ‘simpler’ projects for the smaller-sized interactomes. Such an effort should also be coordinated with an improved mapping of the complete yeast interactome as a model, or test, system. Meantime, there have been since 2005 a few reports of partial, human cancer cell interactomes in the form of predicted maps of human protein interaction networks based on partial data and comparative analysis. Such studies emphasize even further the need and urgency for the complete mapping of several human cancer cell interactomes.
Following the seminal studies of DeRisi et al. in 1996 that utilized cDNA microarray to analize gene expression patterns in human cancer [90], there have been relatively few attempts at deriving hypothetical gene expression patterns in human cancer. The first claim of such an attempt was made by Wachi et al. [262] for genes that were differentially expressed in squamous cell lung cancer tissues from five patients who had undergone surgical removal of the tumor(s) [261]; (cRNA samples were prepared and hybridized to arrays obtained from Affymetrix® (Hg-U133ATM). These authors were able to carry out paired t-test analyses for each individual patient in order to distinguish the genes in which expression levels in their squamous lung cancer cells differed from the paired normal lung tissue (control samples) obtained from the same five individuals. The authors’ prediction methodology will be briefly discussed in the next subsection as some of the details are relevant for the evaluation of these results which were the first to be reported for the (hypothetical) interactome-transcriptome analysis of human cancer cell data for a group of five patients with the same diagnosed form of (lung) cancer, and with the same treatment (tumor removal by surgery).
The hypothetical human protein interaction maps are relatively new endeavors [72,172] perhaps because they are likely to have many false positives, as well as miss a significant fraction of the relevant/real protein-protein interactions. Currently, microarray analysis still suffers inherently from relatively high noise levels and the accompanying information loss (buried in noise); although this inherent noise problem is partially eliminated through multiple replicate analyses, the number of replicates is often limited by the availability and the material cost. Another significant problem of such microarray projects is the huge amount of data that needs to be processed in order to obtain useable information [84].
Analysis of Human Protein-Protein Interactions (HPPI) and integration of array data into a Predicted Protein-Protein Interaction Network (PPIN), (summarized from [261])
Wachi et al. [262] employed for their human cell data analysis a web-presented database (OPHID, April 25, 2005) of predicted interactions between human proteins [72] based on data for human and other four organisms which included the intensely-studied yeast and fruit fly. (OPHID is freely available to academic users at http://ophid.utoronto.ca ). This protein interaction database listed 16,034 known human protein interactions obtained from various public protein interaction databases, as well as 23,889 additional, predicted interactions which are evaluated using protein domains, gene co-expression and Gene Ontology terms. The results can be visualized in OPHID using a customized, graph visualization program. The data comprises literature-derived human PPI from BIND, HPRD and MINT, “with predictions made from Saccharomyces cerevisiae, Caenorhabditis elegans, Drosophila melanogaster and Mus musculus”. The genes in the WYU05 array were matched to those in OPHID using gene symbols and protein sequences. In this manner, 2137 genes in the WYU05 microarray experiments were ‘matched to the protein network from OPHID’. These predictions should, however, be thought of only as ‘hypotheses’ until they are experimentally validated. On the other hand, there is increasing evidence that at least certain PPIs may be conserved through evolution [205,274]. Subsequently, Sharan et al. claimed that about 50% of the protein-protein interactions predicted by using interologs between microorganisms are also experimentally validated [242]. The interologs approach might play therefore a role in the partial validation of the HT-Y2H protein network mapping without, however, necessarily achieving the claimed, global validation of the predicted (hypothetical) interactome.
Differentially expressed genes (DEGs) from squamous cell carcinomas (SCCs) were then identified as discussed above and their connectivity in the network graph was examined to determine their ‘topological’ properties, such as the edge distribution for DEGs in comparison with the surrounding graph sub-network.
Differentially expressed genes –DEG- results for SCC of human lung (summarized from [261])
The genes that are upregulated in SCC were found to exhibit a positive correlation (Pearson’s r-coefficient of 0.82) with the number of edges associated with them (Figure 1a of Wachi et al. [262]), which was interpreted as indicating that DEGs that are upregulated in SCC are also highly connected. However, the downregulated genes were reported also to have a positive correlation (r = 0.75) to connectivity, albeit slightly lower (Figure 1b of Wachi et al. [262]). On the other hand, microarray probe sets that matched the genes in the protein network (n =-2,137) had a negligible correlation coefficient (r =0.06) to link number, proving that the genes on the test microarrays did not contribute to bias in the number of links for DEGs in SCC.
A k-core analysis of DEGs in SCC of the human lung was also carried out (loc. cit.) which was reported to measure “how close are the DEGs to the topological ‘center’ of the human PPI network”. Based on the k-core analysis, it was concluded that: “the upregulated genes are more centrally located in the protein network than the downregulated genes”. If duplicated and validated, such studies would be important as the ‘topological centrality’ of the genes in the interactome was previously reported to be associated with the essential functions of the genes in the yeast [148]. Such essential genes, are lethal when mutated, and also tend to have high connectivity. Moreover, other genes that are not essential in this sense, but provide a vital function in toxin metabolism were reported to have a high number of edges associated with the nodes, and to be less well connected than the essential genes in yeast [224]. Furthermore, a k-core analysis has also been performed on the yeast essential genes and they were reported to be global hubs, whereas the non-essential genes were not hubs [274]. It was also claimed that these essential, global hubs are conserved throughout different species; however, one notes that, thus far, there is insufficient data and evidence to prove this claim, or hypothesis. Nevertheless, one may consider as a ‘working hypothesis’ that “there should be a core set of genes that needs to be maintained throughout the course of somatic evolution in the tumor microenvironment” [259]. This hypothesis is thus consistent with the somatic evolution model of cancer. Such conserved genes might be the ‘essential genes’ in cancer cells, and they may also have somewhat analogous to the global hub, essential genes reported in yeast [272,278]. DEGs would thus be essential for the survival and proliferation of cancer cells in SSC of the human lung, and the upregulated genes would be centrally located in the protein network as well as have higher connectivity, perhaps suggesting their possible essential role(s) in human (SSC) lung cancer. As this is the first report of a predicted/ hypothetical human cancer interactome network one should definitely consider ‘replicating’ the reported studies and also evaluating such potentially important findings in the context of a complete human cancer interactome (differential) analysis. This possibility that DEGs might be essential for the survival and proliferation of cancer cells in SSC of the human lung has much too important consequences to be ignored; therefore, it must be thoroughly investigated and also tested with sufficiently extensive, translational genomics and transcriptional databases that do not seem to be currently available [128]. Additional supporting analyses for this conjecture made by Wachi et al. [262] are further considered in the next subsection.
Cancer proteins and the global topology of the human interactome network
An extensive study of both cancer and non-cancer proteins [127] was integrated into a validated protein-protein interaction (PPI) network, or interactome, of human proteins. In their report, the connectivity properties were investigated for all proteins previously shown to be modified as a result of mutations leading to cancer [86]. A global protein-protein interaction network was then constructed by a homology--based method which is claimed to accurately predict protein-protein interactions. It was then suggested that human proteins that are involved in cancer, or ‘cancer proteins’, exhibit a network topology which is substantially different from that of other proteins which are considered not to be involved in cancer. Notably, increased connectivity was pointed out for cancer proteins involved in the following sub-networks: cell growth and apotosis-related, signal transduction (MAPK, TGF-beta, insulin, T-cell and B-cell receptor, adipocytokine, cytokine-cytokine interaction), cell motility/cytoskeleton, cell communication, adherence junction, focal adhesion, leukocyte migration, antigen processing and folding/sorting/degradation. Furthermore, it was proposed that such observations ‘indicate an underlying evolutionary pressure to which cancer genes, as genes of central importance, are subjected.’ Linking these claims with previous proposals by Wuchty [275] that globally central proteins form an evolutionary backbone of the proteome and are essential to the organism, (and also with the conjecture made by Wachi et al. [262], discussed here). Jonsson and Bates suggested that cancer proteins may generally be older than the non-cancer ones in evolutionary age [3]. Moreover, they also suggested that the somatically mutated cancer proteins may be of somewhat younger evolutionary average age in comparison with those from the germline, as a consequence of the evolutionary selection pressure postulated to affect germline mutated proteins. Note also that the previous study of (SCC) human lung cancer by Wachi et al. also reported in [262] increased interaction connectivity in differentially expressed proteins in human lung cancer tissues.
Epigenetic controls
Upon completion of the US Human Genome Mapping Project and related studies, it became increasingly evident that a sequence of 30,000 or so ‘active’ genes that encode and direct the biosynthesis of specific proteins could not possibly exhaust the control mechanisms present in either normal or abnormal cells (such as, for example, cancer cells). This is even more obvious in the case of developing embryos or regenerating organs. Subsequently, in a 2004 editorial in Nature more than 120,000 genes were suggested to be active in the human genome. Furthermore, specific control mechanisms of cellular phenotypes and processes were recently proposed that involve epigenetic controls, such as the specific acetylation <—> de-acetylation reactions of DNA-bound histones. Such controls intervene from outside the genome but ultimately they also affect gene expression. Therefore, gene profiling techniques would need to be combined with epigenomic tools and analysis in order to gain an improved understanding of functional genomics and interactomics. Epigenomic tools and novel techniques begin to address the complex and varied needs of epigenetic studies, as well as their applications to controlling cell division and growth. Such tools are, therefore, potentially very important in medical areas such as cancer research and therapy, as well as for improving ‘domestic’ animal phenotypes without involving genomic modifications of the organism.
Novel tools in epigenomics: Rapid and ultra-sensitive analyses of nucleic acid –protein interactions
Several novel techniques could also be applied for the highly-selective detection of epigenomic changes in mammalian cells related to diseases such as individual types of cancer [150,215]. Such novel tools are likely to be utilized in a wide range of applications in biotechnology research related to Post-Genomics and Epigenomics. Tumor suppressor genes are transcriptionally silenced by promoter hypermethylation that also appears to lead to alterations in chromatin structure- a possible mechanism for such repression of the suppressor genes. In contrast to the genetic mutation or deletion mechanism of tumor suppressor gene inactivation, epigenetic inactivation of tumor suppressor genes would occur via methylation of specific DNA regions that could be prevented by DNA methyl-transferase or histone deacetylase inhibitors. Aberrant CpG--island methylation has non-random/tumor-type-specific patterns [87]. Such patterns can be identified by employing methylation--specific PCR (MS-PCR; [135]), and can also be employed either for tumor class prediction by microarray-based DNA methylation analysis or for high-throughput microarray-based detection and analysis of methylated CpG islands [276]. Hypermethylation profiling is important for both accurate diagnosis and the development of optimal strategies in cancer therapy. Gene promoter hypermethylation has been reported in both tumors and serum of patients diagnosed with several types of cancer: head and neck cancers, nasopharyngeal carcinoma [273] non-small cell lung cancer [39] gastric carcinoma, liver, prostate, bladder and colorectal cancers [170,190]. Substantial efforts are being made recently for the development of new methods and tools that are capable of sensitive and quantitative DNA methylation analysis, as well as early and accurate diagnosis of cancer. Among such tools are: Fluorescent methylation--specific polymerase chain reaction assay (FMS-PCR; [122]), SNIRF [55,56] (and references cited therein), indocyanine green-labeling (IGL) for human breast carcinomas, ConLight-MSP [6], COBRA [275], Methylation-Sensitive Single Nucleotide Primer Extension (Ms-SnuPE; [123]), DNA microarray sensitive detection by Metal-Enhanced Fluorescence (MASD/MEF; [168,181]), and NIR Fluorescence Microspectroscopy (NIRFMS) for single cancer cell detection [56,58]
Specific molecular markers of cancer [242] hold the promise to identify those molecular signatures that are unique to specific types of cancer, and are essential for the early accurate diagnosis and treatment of cancer. Such novel molecular tools and methodologies could be employed to rapidly and accurately identify molecular signatures of cancer and aging-related diseases in mammalian cells in culture in order to determine how specific epigenomic mechanisms involved in the control of cell division and apoptosis operate throughout the cell cycle. Among the specific epigenomic control mechanisms that one could investigate with such new tools are: CpG-island methylation, p15 (INK4b) and p16 (INK4a) hyper-methylation (in synchronous hepatic carcinoma cells), GSTP1 methylation in non-neoplastic/ synchronous cells, as well as histone-deacetylation and its effects on histone- nucleic acid interactions in stable synchronous cell populations in culture. Both cancer and aging were found to involve DNA methylation of specific genome regions.
Based on the model in (Figure 3), and the data available from the clinical trials and results cited in the Introduction, one has the following potential application strategies to optimizing clinical trials:
A1. One needs to test through quantitative modeling the combined effects of cytostatic anticancer drugs combined in different proportions with the new signaling-based anticancer complement drugs;
A2. The second drug of the SPI type is Cetuximab (C225) that is going to Phase II and III clinical trials.
A3. Pre-clinical studies with C225 showed that Cetuximab results in cell cycle arrest, as well as apoptosis in several types of tumors, and it had synergistic effects with cytotoxic chemotherapy.
Such synergistic effects are readily understood with the modular network in Figure 3 by considering the interconnections between the last two modules at the bottom right of Figure 3.
A4. Flavopiridol applications and potential problems in cancer clinical trials:
a. Flavopiridol causes cell-cycle arrest at G1/S phase transition and G2/M phase transitions and also slows the progression of the cell cycle through the S phase [15,79]
b. The goal was to develop new STKAs that would be similar to flavopyridol, or HMR 1275
c. Some STKAs act as blockers of Cyclin D1, therefore causing cycle arrest by direct transcription repression of cyclin D1 mRNA , and in mantle cell lymphoma, flavopyridol delayed significantly progression of disease in 84% of patients [15,79,97]
d. Cytostatic effects are significant and were observed with the flavopyridol in colorectal and prostate carcinoma xenograft models [86,97]; these findings prompted an evaluation of cyclin-dependent kinases, recognized as responsible for governing the orderly transition from G2 to M phase (CDK1) and G1 to S phase (CDK4 or 6 with CDK2). Indeed, flavopiridol inhibits all CDKs known so far (IC50 100 nM), inhibiting CDK1, CDK2, and CDK4 with a similar potency [79].
e. Undesirable side effects: the risk of thrombosis (among others) makes this drug less than optimal. Preclinical studies of flavopiridol revealed wide differences in growth inhibition between cell types depending on the duration of exposure and concentration of the drug. Significant cytostasis was observed when flavopiridol was administered in protracted fashion to colorectal (colo 205) and prostate (LnCap/DU145) carcinoma xenograft models [97,238]. Shorter “bolus” administration of flavopiridol to a lymphoma/leukemia (HL60) cell line had a higher degree of apoptosis and cytotoxicity [40]. Some cases of partial responses and stable disease have been reported in various phase I studies, but several phase II studies revealed few ‘conventionally defined’ responses in several tumor types, except for the mantle cell lymphoma [22,23,25,86]. In the future, flavopiridol might be tested in combination with other agents, such as: taxol [194], irinotecan [195] and gemcitabine [15], as well as other signal transduction modulators [12,278-279].
f. Despite a number of such issues, it has become apparent that cancer treatments with medicines that control signaling pathways in cancer cells and tumors is a turning point in cancer therapeutics that defines new path for further progress in cancer treatments. One looks forward to future clinical trials in cancer that are optimized, for example, by combining correctly cytotoxic agents with rationally-based signaling strategies that provide the maximum possible benefits to the cancer patients.
g. Assessment and prediction of the effects of therapy with a flavopiridol combination with other SPIs and signal transduction modulators is another example of the potential applications of the modular network model in Figure 3 to achieve rational, individualized cancer therapy in clinical trials. The modular network simulations combined with data obtained from cancer patients undergoing individualized therapy can effectively predict the outcomes of the selected drug combination treatments, thus allowing for the optimization of the cancer clinical trials at the level of individual patients and significantly improve their change of survival through such individualized cancer therapy.
Sample analyses in recent clinical studies have shown that gene expression data can be employed to distinguish between tumor types as well as to predict outcomes. Important, potential applications of such results are individualized human cancer therapy (Pharmacogenomics) and ‘personalized medicine’ that could result in optimized clinical trials in cancer with maximum survival rates of the cancer patients.
Especially relevant are the clinical trials involving ‘inoperable’ lung cancers in which a decade ago survival rates have been unacceptably low and prognosis has been poor. There is a clear need for individualized cancer therapy strategies and pharmacogenomic decisions [284] based on high-throughput microarray information recorded for isolated tumor cell lines from stage I through stage III cancer patients. Gene profiling expression, proteomic, interactomic and tissue array data is essential for improving the survival rate of stage III cancer patients undergoing clinical trials with novel signaling pathway inhibitors/blocker medicines, such as those discussed in some detail in Section 3, (especially subsection 3.2) and Section 6.
Several technologies in conjunction with theoretical models aimed at applications in oncogenesis are currently under development both in the direction of improved detection sensitivity and increased time resolution of cellular events, with the limits of single molecule detection and picosecond time resolution already being reached. Gene expression profiling and epigenomic testing can be carried out with both ultra-sensitive, novel Human and Mouse microarrays. Powerful spectroscopic and microspectrosopic techniques can be then employed for the analysis and further improvement of such tools for the investigation of nucleic ccid-protein interactions.
Novel translational oncogenomics research is rapidly expanding with a view to the application of such new technologies, findings and computational models in both pharmaceutical and clinical areas. Studies of Differential Gene Expression (DFG) in human cancer cell lines are clearly required for developing new strategies for efficient cancer therapies for patients whose tumors have developed resistance to existing therapies.
The urgency for sufficient funding and carrying out complete mappings of highly complex human cancer interactomes in a rationally designed, multi-disciplinary Human Cancer Genomes and Epigenetics (HCGE) Project is pointed out here for the first time.
Such an applied and potentially important project would address the urgent need for rational designs of clinical trials in cancer. The project should be carried out with the help of novel, high-efficiency/low-cost and ultra-sensitive techniques that allowed numerous recent findings in translational oncogenomics. The proposed HCGE project could also greatly benefit from pharmacogenomic approaches and theoretical modeling, computer simulations and predictions of expected, varying structures of human cancer interactomes, The proposed project’s ultimate goal is to optimize clinical trials in cancer and maximize the survival rates of cancer patients treated in such clinical trials.
The author gratefully acknowledges receiving helpful suggestions, pertinent documentation and critical comments from: Dr. Mark Band, Director of the Genotyping/ Transcriptomics Unit at the Keck Center, Dr. Lei Liu, Former Director of the BioInformatics Unit at the Keck Center at UIUC, Professor Schuyler Korban, and Professor James F. Glazebrook of the Mathematics Department at Eastern Illinois University and Adjunct Professor at UIUC.
This research was partially supported by Renessen Co., the IMBA Consortium, and an USDA Hatch Grant No. ILLU-0995362, and AES, at UIUC.