Journal of Plant Biochemistry & Physiology
Open Access
ISSN: 2329-9029
ISSN: 2329-9029
Research Article - (2017) Volume 5, Issue 4
Early diagnosis of fungal pathotypes is one of the most important aspects for plant breeders. It provides a rapid means for selecting the correct mutant lines to ensure durability of disease resistance and also for proper field management practices. Traditional diagnostic tools for pathogens have been based on targeted cultures, PCRbased approaches, and/or phenotypic evaluation of disease response in specific plant genotypes. These methods detect only known pathogenic agents, can introduce bias, and can fail to recognize novel variants or races due to their narrow scope. While enzymatic mismatch cleavage has been described for many plant and animal species, the validate that technique for haploid microorganisms is also needed. In this work, optimized low cost method for pathotyping Fusarium oxysporum f. sp. cubense (Foc), the causal agent of fusarium wilt of banana (Musa spp.), was established using self-extracted nuclease and agarose gel electrophoresis. Gene-specific primers were designed from the whole fungal genome for use in the enzymatic mismatch cleavage on Foc representatives of the major races of major banana producing countries. Gene-specific primer pairs were used to optimize enzymatic mismatch cleavage and polymorphism discovery in two SNF1 and FOW2 genes. The protocol is rapid, inexpensive and can robustly distinguish pathotypes in Foc strains, without high informatics load of DNA sequencing.
Keywords: Single-strand-specific nuclease; Polymorphism discovery; Ecotilling; Banana fusrium wilt; Haploid pathotype
Fungi play a unique role in the Earth’s biomass. Accurate genetic characterization is an important endeavor in many fields including human health, crop sciences and breeding, and for the development of industrial applications. Therefore, emerging, and re-emerging new pathogen pathotypes imperil public health and global food security. Responding to these threats requires improved surveillance and diagnostic systems. Yet, many methods are not suitable for developing countries with varying levels of laboratory infrastructure.
Independent of the host organism, the scale and frequency of emerging diseases has increased with the globalization and industrialization of food production systems [1]. Improved surveillance mechanisms and diagnostic tools are needed to rapidly respond to these emerging threats. However, the time-consuming and tedious protocols required to maintain these pathogens on their hosts have impeded the translation of genomic technologies into surveillance and diagnostics methods. With recent advances in DNA and molecular approaches, Ecotilling method for discovery and characterization of small nucleotide polymorphisms has been widely used for evaluating plant genomes, and to a lesser extent in humans and animals [2,3]. It was originally an adaptation of the Tilling reversegenetics approach applied to Arabidopsis thaliana [4,5]. Reports indicate that Ecotilling is highly efficient for detecting genetic variations associated with target traits for crop improvement. Nieto et al. [6] detected polymorphisms in one haplotype controlling virus susceptibility in a natural melon population. Many polymorphisms in a dozen genes were also identified in wild populations of Populus trichocarpa [7] and in the Musa gene pool [8].
However, despite their potential, genomic tools have not been readily applied to emerging or re-emerging plant pathogens such as the banana wilt (panama disease) pathogen F. oxysporum f. sp. cubense . The threat posed by this new variant, called tropical race 4 (TR4), may be overcome by the introduction of resistant cultivars [9,10]. However, the most effective and environmentally sound means of control is the identification of resistant genotypes or breeding for resistance is a long-term effort. Currently, the only option to control the disease is to avoid or reduce the spread of the pathogen by eradication of infected plants and isolation of infested plantations [11]. This requires sensitive and highly specific diagnostics that enable early detection of the pathogen.
Here, we have developed a low-cost Ecotilling method for polymorphism discovery in a haploid fungus that is suitable for most laboratories. As a proof of principle, Jawhar et al. [12] developed this method for barley spot blotch pathogen (Cochlioblous sativus ) to survey a panel of 50 isolates for allelic variants in different candidate genes. While the methods described in this protocol are straight forward and low cost, a key component of successful application is proper experimental design. Example data and a discussion of experimental design are provided along with a detailed protocol suitable for many plant pathogens species.
Fungal isolates
Four pure DNA single spore strains of Fusarium oxysporum from the rhizosphere of banana plants received from the Department of Plant Pathology, University of Stellenbosch, South Africa were used this work (Table 1). The strains were selected from a global collection of 70 F. oxysporum isolates representing 20 of the 24 VCGs in different major banana growing regions [13].
| Strain | VCG | Cultivar | Country |
|---|---|---|---|
| F1 | 300 | Valery CV-1 | Indonesia |
| F2 | 189 | HarareRPMV40 | Malawi |
| F3 | 180 | C.0121 | Taiwan |
| F4 | 1036 | Latundan | Philippine |
Table 1: Origin of banana fusarium wilt strains used in this study.
Primers design, PCR amplification and heteroduplex formation
PCR primers specific for SNF1 and FOW2 genes (Table 2) were designed from whole genome sequence information from NCBI for F. oxysporum using parameters previously described for Tilling [2]. In order to increase the efficiency of Ecotilling even more, DNA of up to three isolates could be pooled before amplification, just as it is in reverse genetic screens [14]. The PCR reaction was performed in a 20 μl reaction volume containing 1 U AmpliTaq® DNA polymerase, 60 μM dNTPs, 0.75 ng of genomic DNA, and 1.0 μM primers in 1X PCR Buffer (1.5 mM MgCl2, 50 mM KCl, 10 mM Tris HCl, pH 8.3 at RT). After an initial denaturing step at 94°C for 5 min, the DNA was amplified through 20 cycles consisting of denaturing at 94°C for 5 s, annealing at 75 °C for 1 min decreasing by 0.5°C per cycle, and extension at 72°C for 1 min. The samples were then subjected to an additional 30 cycles consisting of denaturing at 94°C for 5 s, annealing at 67°C for 1 min, with a final extension of 5 min at 72°C. Denaturing and re-annealing step is included at the end of the PCR reaction (99°C for 10 min, followed by 70 cycles of 70°C for 20 s decreasing by 0.3°C per cycle) to allow the formation of heteroduplexes if nucleotide polymorphisms are present in the pool. To evaluate the PCR reactions, 4 μL of amplification product were loaded onto a 2% agarose gel and compared to a Precision Molecular Mass Standard (BioRad, Hercules, CA, USA) to determine the approximate concentrations of DNA in each sample, which were then adjusted to ~10 ng/μL in dH2O.
| Primer | Forward primer sequence (5’→3’) | Reverse primer sequence (5’→3’) | Product size |
|---|---|---|---|
| FOW2a | tccagctcccactctgcaacgtc | gcaccgagataacctcaggcaagc | 1211 |
| FOW2b | acgtccaattgacgcgcctgtaa | tcggcttgaatcttgtctggaagca | 1215 |
| FOW2c | cgatatgcccatgtcgcaaacct | caggatcccaagaccggaagacg | 1210 |
| FOW2d | tgagcatgcccaaacaatccctct | ccggaagacgtcagggaacgaac | 1390 |
| FOW2e | gctggccctgggaactcagtttg | tgctgacgtgctagaccaatgttgc | 1254 |
| FOW2f | caggagcgaaggcagacagcaga | gcatgcaatgaatactcgcagcttga | 1371 |
| SNF1a | cgtcgcacccagttcctgacaac | gctcgatgcacagggttcaccac | 1100 |
| SNF1b | ccttgggtcaaggcatcacatcg | gcctgcagggcctcctctacatc | 1247 |
| SNF1c | tgccaagattgcaaagggcacat | tagcgcaacgaaaggctccttgg | 1246 |
| SNF1d | cccgaagtcgatgtgtggagctg | gtcggccatgggctgtctcgtat | 1311 |
| SNF1e | aagaacgacatcgcaccgaatgc | gaaccgtctggcaggttaagaagca | 1312 |
Table 2: Primers designed for Ecotilling assays.
CEL I digestion of the heteroduplexes and agarose gel electrophoresis detection
CEL I was purified from celery to near homogeneity as previously described by Till et al. [15]. The PCR reaction was performed in a 20 μL reaction volume containing containing 6 μl CJE Buffer (10 mM Hepes, pH 7.5, 10 mM MgSO4, 10 mM KCl, 0.002% Triton X-100, and 0.2 μg/ml BSA); 2.4 μl crude celery juice extract (CJE) preparation [2], and 11.6 μl ultrapure water. The digestion was then performed at 45°C for 45 min followed by incubation on ice to stop the reaction. Subsequently, 4 μl of 10 × gel loading buffer was added to each sample.
The digested products were separated on 2% agarose gels with 1 × TAE buffer at 110 V for 50 min, stained in an ethidium bromide buffer (0.5 μg/ml) for 15 min, and then visualized by Gel Doc XR (BioRad Laboratories, Inc.).
Differences between the digested fragments from various DNA pools were assessed visually by recording molecular weights of bands representing cleavage products. To verify the reliability and accuracy of Ecotilling in detecting polymorphisms among isolates, the experiments were repeated twice and resulted similar results.
In this study, A F. oxysporum enzymatic mismatch cleavage strategy was developed for Ecotilling in haploid organism using self-extracted nuclease and agarose gel electrophoresis (Figure 1). A key factor in accurate polymorphism discovery is the development of gene-specific primers. Differences of band patterns among the digested products of different isolates were clearly observed by agarose gel electrophoresis. However, in some pools more than two digestion fragments from one amplified fragment could be detected on agarose gels stained by ethidium bromide.
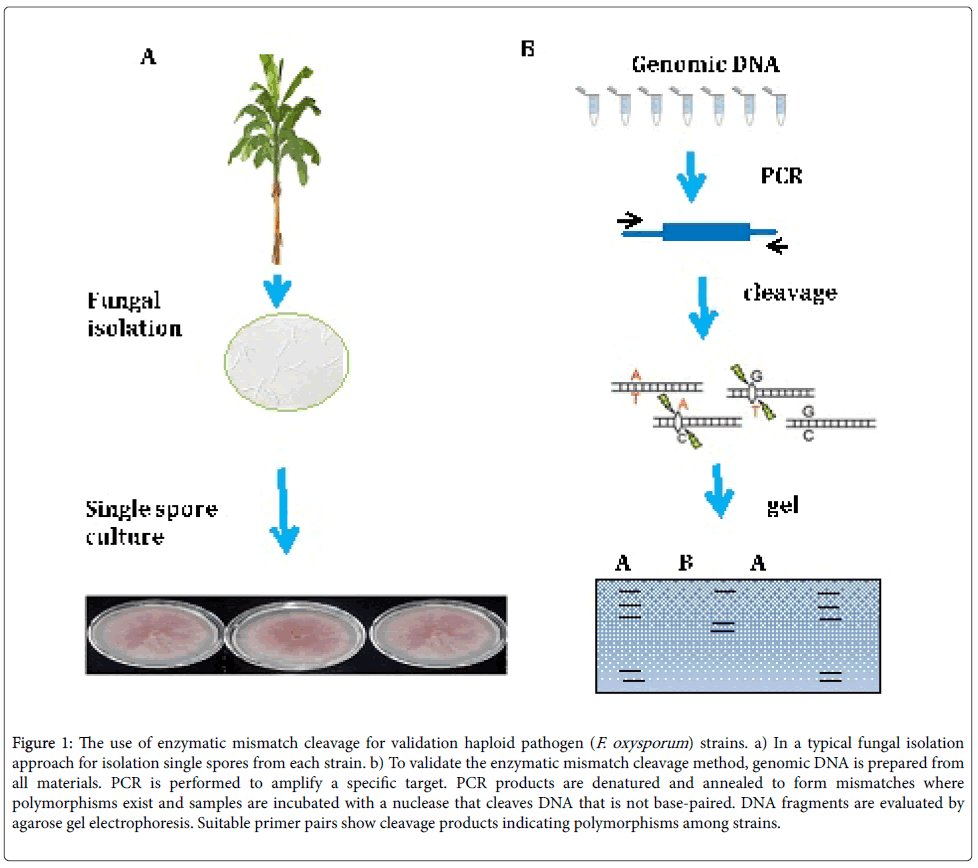
Figure 1: The use of enzymatic mismatch cleavage for validation haploid pathogen (F. oxysporum ) strains. a) In a typical fungal isolation approach for isolation single spores from each strain. b) To validate the enzymatic mismatch cleavage method, genomic DNA is prepared from all materials. PCR is performed to amplify a specific target. PCR products are denatured and annealed to form mismatches where polymorphisms exist and samples are incubated with a nuclease that cleaves DNA that is not base-paired. DNA fragments are evaluated by agarose gel electrophoresis. Suitable primer pairs show cleavage products indicating polymorphisms among strains.
In this protocol, different issues were noticed and should be considered; primer pairs should be selected to different genomic regions that are not genetically linked, and the total number of primer pairs needed depends on the percentage of primer pairs where polymorphisms are observed. Primers were typically designed with the program Primer3 [16] with a melting temperature of 70°C. Additionally, the optimal amount of genomic DNA to be used should be determined empirically. Typically, a PCR amplicon yield of 10 ng/μl is sufficient (Figure 2). The amount of genomic DNA needed to produce this amount can be roughly estimated by size of the genome [2].
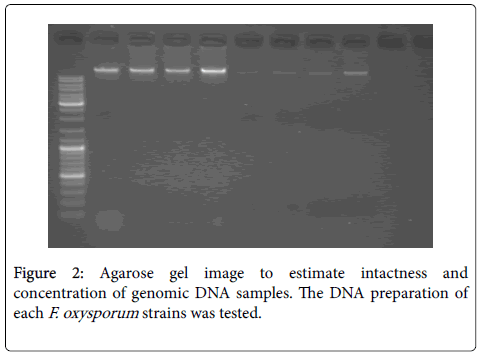
Figure 2: Agarose gel image to estimate intactness and concentration of genomic DNA samples. The DNA preparation of each F. oxysporum strains was tested.
Since this is a haploid organism, individual strains did not form heteroduplexed DNA at the site of a mismatch, therefore, for the success of the method based on using pooling strategy as described in Figure 3. However, to take advantage of agarose gel detection, special attention must be given to the quality and quantity of PCR products. First, it is important that a single specific product is obtained from a PCR reaction since the presence of non-specific products may lead to heteroduplex formation. Non-specific amplification would result in the detection of CEL I-cleaved products even in the control samples. Therefore, it is important that once a polymorphism is detected in a pool, the individuals of the pools be tested separately to confirm that the cleaved products are a result of heteroduplexes formed across members of the pool and not a result of non-specific amplification. Secondly, it is critical to have a high yield of the amplicon. Products at concentrations less than 5 ng/μl would not be detected on regular agarose gels.
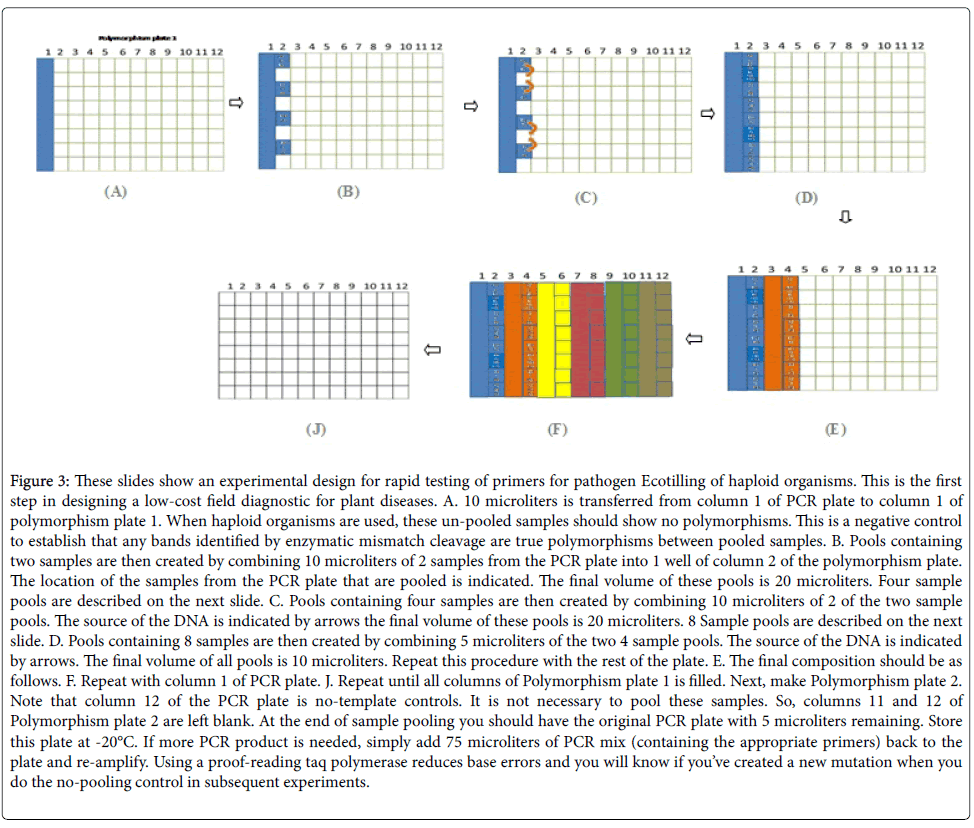
Figure 3: These slides show an experimental design for rapid testing of primers for pathogen Ecotilling of haploid organisms. This is the first step in designing a low-cost field diagnostic for plant diseases. A. 10 microliters is transferred from column 1 of PCR plate to column 1 of polymorphism plate 1. When haploid organisms are used, these un-pooled samples should show no polymorphisms. This is a negative control to establish that any bands identified by enzymatic mismatch cleavage are true polymorphisms between pooled samples. B. Pools containing two samples are then created by combining 10 microliters of 2 samples from the PCR plate into 1 well of column 2 of the polymorphism plate. The location of the samples from the PCR plate that are pooled is indicated. The final volume of these pools is 20 microliters. Four sample pools are described on the next slide. C. Pools containing four samples are then created by combining 10 microliters of 2 of the two sample pools. The source of the DNA is indicated by arrows the final volume of these pools is 20 microliters. 8 Sample pools are described on the next slide. D. Pools containing 8 samples are then created by combining 5 microliters of the two 4 sample pools. The source of the DNA is indicated by arrows. The final volume of all pools is 10 microliters. Repeat this procedure with the rest of the plate. E. The final composition should be as follows. F. Repeat with column 1 of PCR plate. J. Repeat until all columns of Polymorphism plate 1 is filled. Next, make Polymorphism plate 2. Note that column 12 of the PCR plate is no-template controls. It is not necessary to pool these samples. So, columns 11 and 12 of Polymorphism plate 2 are left blank. At the end of sample pooling you should have the original PCR plate with 5 microliters remaining. Store this plate at -20°C. If more PCR product is needed, simply add 75 microliters of PCR mix (containing the appropriate primers) back to the plate and re-amplify. Using a proof-reading taq polymerase reduces base errors and you will know if you’ve created a new mutation when you do the no-pooling control in subsequent experiments.
On the other hand, detecting polymorphimis among F. oxysporum isolates (Figure 4) are not surprising given the scale of this study and what is known about their different origin over several years [13]. These pathotypes tested here were first selected based on morphological and physiological criteria [13,17], and so a high degree of variation may be expected.
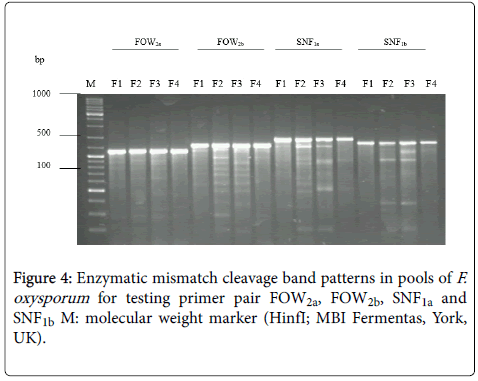
Figure 4: Enzymatic mismatch cleavage band patterns in pools of F. oxysporum for testing primer pair FOW2a, FOW2b, SNF1a and SNF1b M: molecular weight marker (HinfI; MBI Fermentas, York, UK).
This work illustrated the utility of enzymatic mismatch cleavage for Ecotilling in haploid organism F. oxysporum with accuracies similar to those previously achieved for human and plant Ecotilling [2,17]. The protocol could be used to tag strains in gene databases, and for the classification of newly acquired samples. Moreover, this method compares favorably to the most common alternative–full sequencing, since Ecotilling greatly reduces the number of sequences that needs to be determined and is several folds cheap than full sequencing [2]. Additionally, the method is robust and does not require sophisticated instrumentation or costly labeled primers. However, in laboratories without sequencing facilities and where VCG testers of Foc cannot be used because of national quarantine regulations, this low-cost method could be of great value in the characterization of the fusarium wilt pathogen of banana. It is, specifically, its ability to rapidly and accurately detect Foc “tropical” race 4 VCG 300, 194, and 1036 strains in new regions where this pathogen is introduced that could be invaluable in the isolation and management of this most devastating form of F. oxysporum f. sp. cubense .
The authors would like to thank the Director General of AECS and the Head of Molecular Biology and Biotechnology Department for their support. They would also like to thank the Food and Agriculture Organization of the United Nations and the International Atomic Energy Agency through their Joint FAO/IAEA Programme of Nuclear Techniques in Food and Agriculture for supporting the laboratory experiments. We would like also to thank very much to Dr. A. Viljoen (Department of Plant Pathology, University of Stellenbosch, South Africa) for providing us with F. oxysporum strains.
The authors declare that the study involved no conflict of interest from any party.