Journal of Chromatography & Separation Techniques
Open Access
ISSN: 2157-7064
ISSN: 2157-7064
Research Article - (2016) Volume 7, Issue 2
In the present study, we introduced a novel, reliable, sensitive and highly precise capillary zone electrophoresis (CZE) method for simultaneous separation and determination of nalbuphine hydrochloride (NLB) and its related antagonist compounds naloxone hydrochloride (NLX) and naltrexone hydrochloride (NLT) in bulk drug, pharmaceuticals and human biological fluids. The separation conditions using CZE were optimized and the separation process was carried out using fused silica capillary of total and effective lengths 57 and 50 cm, respectively. The applied running buffer was acetate buffer 20 mmol L-1 of pH=3.8 at a potential 15 kV. The detection of samples was carried out using Diode array (DAD) at 230 nm with hydrodynamic injection 6 s, under 70 mbar and capillary cartridge 25°C. The applied internal solution (IS) was phenylethylamine (PEA). The developed method displayed an excellent separation of the investigated drugs with linear concentration ranges of 5-200, 20-240 and 10-280 μg mL-1 for NLB, NLX and NLT, respectively. Low detection limits were recorded as 0.3, 5.0 and 6.5 μg mL-1 while, the quantification limits were 5, 20 and 10 μg mL-1 for NLB, NLX and NLT, respectively. Good correlation coefficients were evaluated at 0.9997, 0.9995 and 0.9996, for the previously selected drugs, respectively. The % RSD was in an acceptable limit indicating high precision. Excellent separation and detection with acceptable results were achieved with respect to migration time, peak area and resolution. The obtained results were statistically evaluated and then compared with those obtained from other published methods. The electrophoretic method was validated in compliance with ICH guidelines.
Keywords: Capillary zone electrophoresis; Nalbuphine hydrochloride; Naloxone hydrochloride; Naltrexone hydrochloride; Pharmaceutical formulations; Biological fluids
The synthetic opioid agonist-antagonist of the phenanthrene series nalbuphine hydrochloride (NLB), is chemically known as 17-(cyclobutylmethyl)-4,5α-epoxymorphinan-3,6α,14-triol hydrochloride (Figure 1a). It is a member of chemically related drugs to the widely used opioid antagonist, naloxone hydrochloride (NLX) and naltrexone hydrochloride (NLT) [1]. The introduced literature survey showed a number of analytical methods concerning the determination of NLB in different matrices including pharmaceutical formulations and biological samples. Among these methods are the chromatographic separation methods, namely high performance liquid chromatography [2,3], thin layer chromatography [4], gas chromatography-mass spectrometry [5] and liquid chromatography coupled with mass spectrometry [6]. Also, other analytical methods such as spectrophotometry [7] and potentiometry [8] have been published. NLX is a narcotic antagonist drug used for treatment of overdoses of narcotic medications [9]. It is a member of isoquinoline compounds (Figure 1b). Many analytical methods have been reported for the detection of NLX mainly by high performance liquid chromatography [10,11], high performance liquid chromatography coupled with mass spectrometry [6,12-15], chemiluminescence [16] and p otentiometry [17].

Figure 1: Chemical structures of nalbuphine hydrochloride, naloxone hydrochloride and naltrexone hydrochloride.
NLT, (Figure 1c) is an opioid receptor antagonist which has a long action to block the subjective effect of alcohol dependence and opioid dependence [18]. The chemical name of naltrexone is 17-(cyclopropylmethyl)-4,5-epoxy-3,14-dihydroxymorphinan-6- one,(5α)-hydrochloride [19]. Many analytical methods have been reported for determination of naltrexone hydrochloride including RP-high performance liquid chromatography [20,21], liquid chromatography-mass spectrometry [22], spectrophotometry [23], spectrofluorimetry [24] and potentiometry [25].
In recent years, much attention has been undertaken for using CZE in wide fields of chemical analysis [26,27]. Although, the chromatographic separation methods such as LC-MS has a high sensitivity and selectivity for drug analysis, the proposed CZE method offered many advantages, including cost benefit technique, improved separation speed and less solvent consuming rather than other chromatographic separation techniques.
Regarding to the studied drugs NLB, NLX and NLT no spectroscopic analysis methods have been reported yet for simultaneous separation and determination of the selected drugs. Therefore, the aim of the present study is to develop a novel, rapid, sensitive and reliable method for simultaneous separation and estimation of NLB and its related antagonist compounds NLX and NLT. The described method was applied in the detection of the investigated drugs and further extended to be validated according to ICH guidelines [28].
Instrumentation and software
The electrophoretic separation was done using (PrinCE 770-Technology) instrument, which comprises a fused silica capillary with a total and effective lengths of 57 and 50 cm, respectively and 75 μm i.d. It was connected with diode-array detector (DAD) for peak detection. The CZE PrinCE 770-Technology system was equipped with Autosampler and a thermostated column cartridge was connected to adjust the temperature. Also, it has a high voltage built in power supply. Automated, controlled PC for electrophoretic separation was applied using a WinPrinCE-770, DA × 3D software for data acquisition. HANNA 211-pH meter connected with Ag/AgCl reference electrode was used for adjusting the electrolyte pH. The HPLC-DAD detection was carried out using Agilent 1200 series (Agilent Technologies, Santa Clara, California, USA), coprises of pump, vacuum degasser and diode array detector. The column used Zorbax SB-C18 (4.6 × 250 mm, 5.0 mm particle size). The system was PC controlled and the computer was loaded with Agilent ChemStation Software.
Materials and reagents
Analytical grade of all materials and reagents were used and all solvents were of HPLC spectroscopic grade. The electrophoretic separation was performed using acetate buffer pH=3.8 (freshly prepared using 0.1 mol L-1 acetic acid and 0.1 mol L-1 sodium acetate). Phosphate buffer using mono and dibasic hydrogen phosphate solutions with pH 5.8 - 8.0, glacial acetic acid ≥ 99.0%, phenylethylamine hydrochloride (PEA) ≥ 99.0%, orthophorphoric acid and ammonium acetate 99.0% were purchased from (Sigma-Aldrich, Hamburg, Germany). Also, acetonitrile, methanol, ethanol and isopropanol were supplied from (BDH, Philadelphia, USA). Zinc sulfate ≥ 99.0%, sodium hydroxide ≥ 99.0%, sodium acetate, boric acid and sodium dihydrogen phosphate ≥ 90.0 % were purchased from (WinLab, East Midlands, UK). Pure grade of nalbuphine hydrochloride was kindly provided from (Amoun Pharmaceutical Co., Cairo, Egypt). While, naloxone hydrochloride and naltrexone hydrochloride were supplied by (Bristol Myer Squibb Co., Giza, Egypt). Nalufin® ampoules 20 mg mL-1 of NLB, Vivitrol vials 380 mg/vial of NLT and Narcan® ampoules, 400 μg mL-1 of NLX were purchased from local drug stores. Deionized water was used throughout the experiments. The urine samples were provided from healthy volunteers. Informed consent was obtained from all of them prior to the start of the study. The study was approved by the Medical Ethics Committee in the College of Medicine, King Saud University. Commercial sources were supplied the human serum samples (Multi- Serum Normal, Randox Laboratories, Crumlin, Antrim, UK).
Preparation of analytical samples
Standard solutions: Freshly, 300 μg mL-1 stock solutions of NLB, NLX and NLT were prepared by dissolving 30 mg of each pure drug in 100 mL deionized water. Phenylethylamine hydrochloride 100 μg mL-1 was applied as (IS) and prepared by dissolving 10 mg of PEA in 100 mL deionized water. Deionized water was used for serial dilution of daily working solutions.
Preparation of authentic mixtures: The electrophoretic analysis was performed using a working solution 100 μg mL-1 of each drug in the presence of 1.0 mL of 100 μg mL-1 IS. Aliquots of NLB, NLX and NLT standard solutions in the final concentration ratios of 1:1:1, 1:2:2, 1:4:4, 1:6:6, 1:8:8 and 1:10:10 (w/w) respectively were mixed and subjected to analysis. The regression equation was applied to calculate the percentage recoveries of each drug.
Preparation of Nalufin, Narcan and Vivitrol injection solutions: The standard Nalufin injection solution was prepared by transferring the content of two ampoules Nalufin ampoules into a 100-mL volumetric flask and diluted with deionized water to obtain a solution containing 400 μg mL-1 NLB. In case of Narcan ampoules the content of ten ampoules was diluted with deionized water in 20-mL volumetric flask to obtain 200 μg mL-1 NLX. A stock solution of the NLT was prepared by transferring the content of one vial of Vivitrol injection into a 100-mL volumetric flask. Then, it was diluted with deionized water to obtain a solution containing 3800 μg mL-1 of NLT. Working solutions were prepared in the ranges of 50-200, 100-240 and 50-280 for NLB, NLX and NLT, respectively. The electrophoretic detection of the investigated drugs was carried out in the presence of 1.0 mL of 100 μg mL-1 of IS.
Preparation of biological samples
The proposed CZE method was successfully applied for determination of NLB, NLX and NLT in human serum and urine. Spiking technique method was used for detection of the investigated drugs in biological fluids. 1.0 mL of human serum was spiked with different aliquots of each drug. Serum deprotination was performed by adding 1.0 mL of acetonitrile, 0.1 mL of NaOH (0.1 mol L-1) followed by 1.0 mL of ZnSO4.7 H2O (5.0% w/v). The prepared solution was centrifuged at 3500 rpm for 30 min. Then the clear layer was filtered using 0.5 Milli-pore a membrane filter. The human urine samples were collected from healthy volunteers, 5.0 mL of urine was spiked with accurately measured aliquots of the investigated drugs separately. Then the solutions were diluted with deionized water no further treatment was required. Working solutions were obtained by serial dilutions with the same solvent.
Electrophoretic conditions
In the present study, a new developed CZE method for simultaneous separation and determination of NLB, NLX and NLT was applied. CZE separation of the selected drugs was performed under optimum conditions using 20 mmol L-1 acetate buffer pH=3.8. The capillary should be conditioned before carrying the separation process using 0.1 mol L-1 sodium hydroxide for 2 min followed by deionized water for 2 min and then equilibrated with running electrolyte for 5 min. Hydrodynamic injection of the samples was carried out under applied voltage 15 kV, capillary cartridge temperature of 25°C and applied pressure of 70 mbar for 6 s. To ensure the separation reproducibility throughout the experiment, the capillary was replenished by 0.1 mol L-1 sodium hydroxide for 5 min, deionized water for 5 min and running buffer electrolyte for 10 min.
Calibration curve
The calibration curves of the investigated drugs were plotted using different concentration ranges of 5-200, 20-240 and 10-280 μg mL-1 for NLB, NLX and NLT, respectively. The obtained data was recorded in the presence of 1.0 mL of 100 μg mL-1 of PEA as IS. The sample injection was triplicated for each concentration. The peak area ratio of each concentration with respect to the IS vis. corresponding standard concentration was plotted to obtain the calibration graphs. Then, the corresponding regression equations were derived.
The developed CZE method was employed for simultaneous separation and determination of a mixture of the investigated drugs NLB, NLX and NLT of (100:40:40) μg mL-1, respectively, in the presence of 100 μg mL-1 PEA as IS. Figure 2 showed the typical electropherogram for the laboratory mixture of the selected drugs. It was found that under optimum conditions the described method exhibited excellent separation of NLB, NLX and NLT at retention times 5.19, 4.21 and 4.55 min, respectively. The proposed method was encouraged to determine the selected drugs with high sensitivity and accuracy in dosage forms and biological fluids.

Figure 2: Typical electropherogram of a mixture of NLB (100 μg mL-1), NLX (40 μg mL-1), NLT (40 μg mL-1) and 100 μg mL-1 IS.
Optimization of CZE conditions
The key strategy to optimize the CZE separation, the degree of ionization of the investigated drugs and their electrophoretic mobility is the type, pH and the concentration of the running buffer solution used. The selected drugs (NLB, NLX and NLT) have pka 8.71, 7.94 and 8.13, respectively. By using an acidic buffer these compounds can be positively changed and separated using CZE technique.
Selection of running buffer solution
Owing to the selection of the suitable running buffer electrolyte considered as one of the most important parameters in electrophoretic separation, the concentration range of 5-50 mmol L-1 of each phosphate, acetate and borate buffer solution was investigated. Under constant instrumental conditions (applied voltage, applied pressure, injection time, temperature and wavelength, etc.), each selected buffer was tested. The recorded results indicated that the most reasonable resolution, signal intensity and migration time was achieved by using acetate buffer solution. Therefore, it was selected for further studies.
Effect of pH
The separation in CZE is very sensitive to pH changes rather than in HPLC. Therefore, small change can greatly affect the separation. Also, one of the typical or the most common buffer used in CZE separation is acetate buffer pH 3.8. The pH value of the running buffer was investigated to ensure excellent separation of the selected drugs. The mobility (ueff) curve of NLB, NLX and NLT and IS was plotted. As shown in Figure 3, the investigated drugs were separated using acetate buffer of pH value 3.8. At pH less than 3.0 no possible separation was obtained, this may be attributed to the interaction of the drugs with internal capillary wall. Therefore, the pH interval 3-7 was tested in the preliminary studies.

Figure 3: Effect of buffer pH on the migration time: optimum conditions 20 mmolL-1 acetate buffer pH=3-7, injection time 6 s, 25°C, 230 nm, 15kV and 50 μg mL-1 of each of the tested drugs and 100 μg mL-1 of IS
Effect of running buffer concentration
The relation of the buffer concentration and the separation process in CZE is very important and the mechanism of action based on the stacking phenomenon which explained by keeping the conductivity of the sample less than the conductivity of the buffer. Also, there are other factors which affect the separation and they are related to the buffer concentration such as EOF. The increasing of buffer concentration will increase the separation process but also it will cause a decrease in the EOF through the capillary. The produced current as well as the electroosmotic flow (EOF) in the capillary was greatly influenced by the concentration of the running buffer solution which applied during the electrophoretic separation. Therefore, to investigate the effect of the acetate buffer concentration on the electrophoretic separation of the selected drugs, 5-50 mmol L-1 of acetate buffer solutions were tested. Figure 4, demonstrated that a high separation performance was obtained under constant conditions (pH 3.8, 70 mbar, 15 kV, 25°C) by using 20 mmol L-1 acetate buffer.

Figure 4: Effect of buffer concentration on the migration time: optimum conditions 5-50 mmol L-1 acetate buffer pH=3.8, injection time 6 s, 25°C, 230 nm, 15 kV and 50 μg mL-1 of each of the tested drugs and 100 μg mL-1 of IS.
Effect of additives and organic modifiers
The effect of some additives to the system, electrolyte was tested by adding 5-25 mol L-1 sodium dodecyl sulfate (SDS) and betacyclodextrin (β-CD). The obtained results revealed that no significant improvement in the separation of the investigated drugs was recorded by adding (β-CD). On the other hand, it was found that the addition of SDS in the level above the critical micelle concentration promotes the aggregation of the surfactant molecules, hence causes, interaction of hydrophobic molecules leading to change in the mobility of the analytes [29]. Therefore, the experiment was carried out in the absence of SDS or (β-CD). Moreover, one of the most critical parameters which should be investigated is the addition of organic modifiers such as methanol (MeOH), ethanol (EtOH), isopropanol (IPA) and acetonitrile (ACN) due to their effect on the electroosmotic mobility, zeta potential and dielectric constant of the CZE. To our knowledge, the analytes move across the capillary under the effect of electroosmotic and electrophoretic forces. The velocity of solute was calculated using the algebraic sum of electrophoretic velocity (Vef) and electroosmotic velocity (Veo) according to the following equation:
Vnett=Vef+Veo=[Dζef/4πη+Dζeo/4πη] E
Where, η is the medium viscosity, ζ is the Zeta potential and E is the strength of the electric field. Moreover, the increase of proportion of organic modifiers added causes a significant decrease in zeta potential and dielectric constant [30].
To investigate the effect of adding organic modifiers to the buffer electrolyte, different % (10-70 v/v) of each organic modifier (MeOH, EtOH, IPA and ACN) was added. As shown in Figure 5, it was found that increasing the proportion of the organic modifier caused a considerable increase in the migration time and the viscosity of the running buffer, but no significant improvement in drugs separation was recorded by adding the organic modifiers. Therefore, the electrophoretic analysis of the investigated drugs was performed without adding organic modifiers and this considered as an Eco friendly method.

Figure 5: Effect of percentage (v/v) organic modifiers on the viscosity of the running buffer solution.
Effect of applied voltage
Under optimum conditions the effect of the applied voltage was tested by performing several runs with gradual increase of the applied voltage from 10-35 kV. For our knowledge, direct relationship was obtained between the efficiency of the resolution (Rs) of analysis and the applied voltage [31]. Therefore, the resolution efficiency was increased by increasing the applied voltage in the range of 15-25 kV. While, it was noticed that excessive Joule heat was generated with further increase of the applied voltage more than 30 kV, which gave a significant decrease in the Rs efficiency of the capillary. As indicated in Figure 6, 15 kV was selected to be suitable for further detection.

Figure 6: Effect of different voltages on separation of NLB (100 μg mL-1); NLX (40 μg mL-1), NLT (40 μg mL-1) and 100 μg mL-1 IS; running buffer; 20 mmol L-1 acetate buffer; injection 70 mbar for 6 s; separation voltage (15-45 kV); capillary temperature 25°C and DAD detection at 230 nm.
Effect of capillary cartridge temperature
Due to the influence of the temperature of the capillary on the EOF and electrophoretic mobility, the capillary cartridge temperature should be controlled and optimized. The temperature of the capillary cartridge was investigated in the range of 25-35°C. It was obtained that excellent separation with good resolution and short migration time was recorded at 25°C.
Selection of injection time
In electrophoretic analysis, the peak width and peak height were affected by injection time. So, the samples of the investigated drugs were hydrodynamically injected under 70 mbar and injection time varied from 2-10 s. It was found that after 6 s peaks deformation was seen. Therefore, 6 s was selected for further studies.
Selection of internal standard
The role of IS in electrophoretic separation is to set off the injection errors, improves the performance of the electrophoretic separation and detection. Also, it lowers the migration time of the separation process. 9.78 is the pKa value of phenylethylamine and its molecular weight is less than the investigated drugs. It will be expected that under acidic conditions the IS positively charged and eluted before the investigated drugs.
Selection of the detection wavelength
The suitable wavelength for electrophoretic separation and detection of the investigated drugs should be optimized. The electrophoretic separation was performed at 190-400 nm. It was found that excellent separation and best signal to noise ratio were achieved at 230 nm.
Method Validation
According to ICH guidelines [28], the developed CZE method was validated with respect to system stability, linear concentration range, accuracy, precision, specificity, limits of detection and quantification and robustness.
Linearity
In order to establish the linear relationship of the developed CZE method, the peak area ratio of the studied drug/IS as a function of drug concentration was plotted. The developed method was established linear relationships at concentration ranges of 5-200, 20-240 and 10- 280 μg mL-1 for NLB, NLX and NLT, respectively. Good correlation coefficients (r) were found to be 0.9997, 0.9994 and 0.9996 with regression equations YNLB=0.0024x+0.4718, YNLX=0.0018x+0.3183 and YNLT=0.0011x+0.3898 for the three mentioned drugs, respectively (Figure 7).

Figure 7: Typical calibration graphs of the investigated drugs (a) NLB, (b) NLX and (c) NLT using CZE method under the optimum conditions: running buffer; 20 mmol L-1 acetate buffer; injection 70 mbar for 6 s; separation voltage 15 kV; capillary temperature 25°C and DAD detection at 230 nm.
Limit of detection (LOD) and limit of quantification (LOQ)
The guidelines ICH Q2 (R1) were used for calculation of LOD and LOQ according to the following equations: LOD=3.3 Sa/b and LOQ=10 Sa/b. Where, Sa is the standard deviation of the intercept and b is the slope. Under optimum electrophoretic conditions it was found that the investigated drugs were detected with LOD of 0.3, 5.0 and 6.5 μg mL-1 and quantification limits of 5, 20 and 10 μg mL-1 for NLB, NLX and NLT, respectively (Table 1). Also, Figure 8, showed the electropherogram of blank sample, LOQ and LOD solutions.

Figure 8: Comparative electropherograms of blank sample solution, LOQ of a mixture of NLB (100 μg mL-1), NLX (40 μg mL-1), NLT (40 μg mL-1) and 100 μg mL-1 IS and LOD of the same sample solution.
| Parameter | NLB | NLX | NLT |
|---|---|---|---|
| Linearity range (µg mL-1) | 5-200 | 20-240 | 10-280 |
| Regression equation | y=0.0024x+0.4718 | y= 0.0018x+0.3183 | y=0.0011x+0.3898 |
| Correlation coefficient (r) | 0.9997 | 0.9995 | 0.9996 |
| Standard deviation of residuals, Sy/x | 0.0041 | 0.0042 | 0.0062 |
| Standard deviation of intercept, Sa | 0.0002 | 0.0025 | 0.0015 |
| Standard deviation of slope, (Sb) | 0.0002 | 0.0001 | 0.0001 |
| Limit of detection (LOD) | 0.3 | 4.6 | 6.5 |
| Limit of quantification (LOQ) | 5 | 20 | 10 |
| %RSD | 0.2 | 0.2 | 0.7 |
| %Error* | 0.0001 | 0.0001 | 0.0064 |
*% Error was calculated by SD/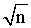
Table 1: Critical performance data of the determination of the investigated drugs using CZE method.
Accuracy and precision
The accuracy of the developed CZE method for separation and determination of NLB, NLX and NLT was evaluated as mean percentage recoveries (n=6) using standard solutions of the investigated drugs. The obtained results were statistically treated and compared with those obtained from other reported methods, high-performance liquid chromatography determination of nalbuphine hydrochloride using C18 column and mobile phase sodium acetate buffer pH 5.5: acetonitrile (40:60 v/v), flow rate 1.0 mL/min and UV-detection at 210 nm [2], high performance liquid chromatography determination of NLX using C18 column, mobile phase 10 mmol L-1 potassium phosphate buffer pH 6.0 with orthophorphoric acid: acetonitrile (17:83 v/v), flow rate 1 mL/ min and UV detection at 210 nm [10] high performance liquid chromatography determination of NLT which based on the using of C18 column, mobile phase ammonium acetate buffer pH 5.8 : acetonitrile (60:40 v/v), flow rate 1 mL/min and UV detection at 220 nm [21] for NLB, NLX and NLT, respectively). The obtained data were presented in Table 2 which indicated that no significant difference was recorded.
| Drug | Proposed CZE method | Reference method | Ref. [2] | |||||
|---|---|---|---|---|---|---|---|---|
| NLB | Taken µg mL-1 | Found µg mL-1 | % Recovery | Taken µg mL-1 | Found µg mL-1 | % Recovery | ||
| 5 50 100 140 160 200 |
4.99 49.85 98.88 139.47 159.53 199.74 |
99.80 99.70 98.88 99.62 99.70 99.87 |
2 4 6 8 10 15 |
1.99 3.98 5.87 8.00 9.99 14.88 |
99.50 99.50 97.83 100.00 99.90 99.20 |
|||
| Mean ± SD n Variance **%SE t-test F-test |
99.60 ± 0.4 6 0.16 0.16 0.845(2.228)* 3.06(5.05)* |
99.32 ± 0.7 6 0.49 0.29 |
||||||
| NLX | 20 60 100 140 160 180 |
19.77 59.28 98.65 139.47 158.98 179.85 |
98.85 98.80 98.65 99.99 99.36 99.92 |
10 20 40 60 80 100 |
10.00 19.99 39.85 59.78 79.63 98.85 |
100.00 99.95 99.62 99.63 99.53 98.85 |
Ref [10] | |
| Mean ± SD n Variance **%SE t-test F-test |
99.26 ± 0.6 6 0.36 0.24 1.144(2.228)* 2.25(5.05)* |
99.59 ± 0.4 6 0.16 0.16 |
||||||
| NLT | 10 50 80 100 150 200 |
10.00 49.58 79.89 99.85 149.57 199.74 |
100.00 99.16 99.86 99.85 99.71 99.87 |
12 16 20 24 28 32 |
11.95 15.99 19.68 23.98 28.00 31.67 |
99.58 99.93 98.40 99.92 100.00 98.97 |
Ref [21] | |
| Mean ± SD n Variance **%SE t-test F-test |
99.74 ± 0.3 6 0.09 0.12 1.006(2.228)* 4.00(5.05)* |
99.47 ± 0.6 6 0.36 0.24 |
||||||
*Figures in parentheses are the tabulated values of t-and F- testes at 95% confidence limit [32]; **%SE= SD /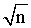
Table 2: Analytical results of the determination of NLB, NLX and NLT in bulk powder using CZE method and reference methods.
To prove the precision of the developed CZE method the intermediate precision was applied using inter-day and intra-day assay and the % RSD was evaluated. As listed in Table 3, the obtained data provided a good precision of the developed CZE method.
| Parameter | NLB | NLX | NLT | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Taken µg mL-1 | Taken µg mL-1 | Taken µg mL-1 | |||||||
| Inter-day | 5 | 50 | 150 | 80 | 160 | 200 | 50 | 200 | 240 |
| % Found | 99.5 | 99.68 | 99.26 | 99.75 | 99.23 | 100 | 99.82 | 99.96 | 99.84 |
| 100 | 99.47 | 99.86 | 100 | 99.74 | 99.48 | 99.26 | 99.24 | 99.12 | |
| 99.87 | 99.12 | 99.98 | 99.52 | 99.24 | 99.86 | 99.42 | 99.87 | 99.63 | |
| Mean | 99.79 | 99.43 | 99.7 | 99.76 | 99.4 | 99.78 | 99.5 | 99.69 | 99.53 |
| ± SD | 0.26 | 0.28 | 0.39 | 0.24 | 0.29 | 0.27 | 0.29 | 0.39 | 0.37 |
| %RSD | 0.26 | 0.28 | 0.39 | 0.24 | 0.29 | 0.27 | 0.29 | 0.39 | 0.37 |
| %SE | 0.15 | 0.16 | 0.23 | 0.14 | 0.18 | 0.16 | 0.17 | 0.23 | 0.21 |
| Intra-day | 5 | 50 | 150 | 80 | 160 | 200 | 50 | 200 | 240 |
| % Found | 100 | 99.96 | 100 | 99.25 | 99.64 | 99.26 | 100 | 99.27 | 99.32 |
| 99.98 | 99.98 | 100 | 99.69 | 99.87 | 99.78 | 99.65 | 99.65 | 99.89 | |
| 99.97 | 99.99 | 99.58 | 99.97 | 99.27 | 100 | 99.87 | 99.97 | 99.47 | |
| Mean | 99.98 | 99.98 | 99.86 | 99.64 | 99.59 | 99.68 | 99.84 | 99.63 | 99.56 |
| ± SD | 0.02 | 0.02 | 0.24 | 0.36 | 0.3 | 0.38 | 0.18 | 0.35 | 0.29 |
| %RSD | 0.02 | 0.02 | 0.24 | 0.36 | 0.3 | 0.38 | 0.18 | 0.35 | 0.29 |
| %SE | 0.01 | 0.01 | 0.14 | 0.21 | 0.17 | 0.22 | 0.1 | 0.2 | 0.17 |
Table 3: Analytical data of inter-day and intra-day precisions for the determination of NLB, NLX and NLT using CZE method.
Robustness of the developed method
To evaluate the robustness of the described CZE method, minor deliberated changes in method parameters were introduced. These parameters include the change in the running buffer solution pH (3.8 ± 0.2), the concentration of the running buffer (20 ± 5 mmol L-1), the temperature of capillary cartridge 25 ± 2°C, injection time 6 ± 1 s and applied voltage 15 ± 2 kV with only one parameter at a time was changed. As reported in Table 4, the peak area ratio and the migration time were not significantly affected by these changes.
| Parameter | Migration time, min | Peak area ratios | ||||
|---|---|---|---|---|---|---|
| NLB | NLX | NLT | NLB | NLX | NLT | |
| Standard | 5.19 | 4.21 | 4.55 | 0.71 | 0.57 | 4.35 |
| Acetate buffer pH 3.6 4.0 |
5.24 5.12 |
4.14 4.22 |
4.62 4.46 |
0.72 0.71 |
0.57 0.57 |
4.35 4.35 |
| Acetate buffer concentration, mmolL-1 15 25 |
5.15 5.30 |
4.12 4.23 |
4.35 4.62 |
0.70 0.71 |
0.55 0.57 |
4.34 4.35 |
| Injection time, s 5 7 |
5.02 5.17 |
4.20 4.16 |
4.56 4.48 |
0.69 0.70 |
0.56 0.57 |
4.33 4.35 |
| Applied voltage, kV 13 17 |
5.34 5.13 |
4.50 4.30 |
4.68 4.66 |
0.71 0.74 |
0.62 0.57 |
4.37 4.36 |
| Capillary cartridge temperature, °C 23 27 |
5.36 5.20 |
4.32 4.24 |
4.69 4.63 |
0.71 0.69 |
0.63 0.64 |
4.37 4.38 |
Table 4: Robustness data using 100 µg mL-1 NLB, 40 µg mL-1 of NLX and 40 µg mL-1 in the presence of 100 µg mL-1 IS.
Specificity
To discriminate the investigated drugs from all other interfering species, standard drugs and some common spiking interfering species such as citric acid, sodium citrate dehydrate, sodium chloride, methylparaben and carboxymethylcellulose sodium salt were used. The peak purity against the pure standard drugs was recorded using a diode array detector and prinCE-770 DA × 3D software. The obtained results demonstrated that no separation peaks were detected at the retention time of each investigated drug and the IS at the lower limit of quantification. Therefore, the developed CZE method for determination of NLB, NLX and NLT was selective and specified for separation and detection of the previously mentioned drugs.
Analytical Applications
Quantification of nalbuphine, naloxone and naltrexone hydrochloride in authentic mixture
The developed electrophoretic method was employed for simultaneous separation and estimation of the tested drugs in their laboratory authentic mixture. Triplicate injection was applied for each sample. The percentage recoveries of each concentration were calculated and found to be 99.00 ± 0.8, 99.65 ± 0.4 and 99.62 ± 0.2 for NLB, NLX and NLT, respectively. The obtained results as reported in Table 5 were statistically treated using student's t-test and variance F-test [32], and then compared with data obtained from other reported methods [2, 10 and 21 for the three drugs, respectively]. Regarding to the accuracy and precision good agreements and no significant difference was recorded. Furthermore, the three drugs were determined in pharmaceutical formulations, human serum and urine.
| Ratio NLB:NLX:NLT % w/w |
Taken μg mL-1 |
NLB | Reference method [2] Taken 2-12 μg mL-1 |
NLX | Reference method [10] Taken 10-100 μg mL-1 |
NLT | Reference method [21] Taken 12-32 μg mL-1 |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Found μg mL-1 |
% Recovery | Found μg mL-1 |
% Recovery | Found μg mL-1 |
% Recovery | Found μg mL-1 |
% Recovery | Found μg mL-1 |
% Recovery |
Found μg mL-1 |
% Recovery |
||
| 1:1:1 1:2:2 1:4:4 1:6:6 1:8:8 1:10:10 |
20:20:20 20:40:40 20:80:80 20:120:120 20:160:160 20:200:200 |
19.98 19.87 19.52 19.69 19.84 19.88 |
99.9 99.4 97.6 98.5 99.2 99.4 |
1.97 3.99 5.87 7.95 9.86 11.96 |
98.5 99.8 97.8 99.4 98.6 99.7 |
19.85 39.57 79.99 120.00 159.96 199.89 |
99.3 98.9 99.9 100.0 99.9 99.9 |
9.99 19.98 39.86 59.87 79.86 98.95 |
99.9 99.9 99.7 99.8 99.8 98.9 |
19.87 39.74 79.63 119.82 159.99 199.25 |
99.4 99.4 99.5 99.9 99.9 99.6 |
11.98 15.86 20.00 23.87 27.93 32.00 |
99.8 99.1 100.0 99.5 99.8 100.0 |
| Mean ± SD Variance **%SE t-test F-test |
99.00 ± 0.8 0.64 0.33 0.068(2.228)* 1.31(5.05)* |
98.97 ± 0.7 0.49 0.29 |
99.65 ± 0.4 0.16 0.16 0.100(2.228)* 1.78(5.05)* |
99.67 ± 0.3 0.09 0.12 |
99.62 ± 0.2 0.40 0.08 0.555(2.228)* 4.44(5.05)* |
99.70 ± 0.3 0.09 0.12 |
|||||||
* Figures in parentheses are the tabulated values of t-and F- testes at 95% confidence limit [32]. ** %SE= SD/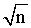 .
.
Table 5: Analytical results of the determination of NLB, NLX and NLT in authentic mixture using CZE method and reference methods.
The evaluated data were summarized in Tables 6 and 7. Firstly, in pharmaceutical preparations, percentage recoveries were calculated and it was found to be 99.43 ± 0.5, 99.36 ± 0.6 and 99.73 ± 0.3. While, in biological fluids the recorded results in human serum were 99.21 ± 0.6, 99.45 ± 0.6 and 99.24 ± 0.7 and in human urine were 99.33 ± 0.5, 99.03 ± 0.6 and 99.50 ± 0.4 for NLB, NLX and NLT, respectively.
| Drug | Proposed CZE method | Reference method | Ref. [2] | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NLB®Nalufin 50- 200 µg mL-1 |
Taken µg mL-1 |
Found µg mL-1 |
% Recovery |
Taken µg mL-1 |
Found µg mL-1 |
% Recovery |
||||||
| 50 100 140 160 180 200 |
49.15 99.82 139.36 159.12 179.75 199.32 |
98.30 99.82 99.54 99.37 99.86 99.66 |
2 4 6 8 10 15 |
2.00 3.96 5.87 8.00 9.96 14.99 |
100.0 99.0 97.8 100.0 99.6 99.9 |
|||||||
| Mean ± SD n Variance %SE t-test F-test |
99.43 ± 0.5 6 0.25 0.20 0.129(2.228)* 2.56(5.05)* |
99.38 ± 0.8 6 0.64 0.33 |
||||||||||
| NLX®Narcan 100-240 µg mL-1 |
100 140 160 180 200 240 |
98.35 139.12 158.53 179.76 199.45 239.47 |
98.35 99.37 99.08 99.87 99.73 99.78 |
10 20 40 60 80 100 |
9.99 19.89 39.95 59.85 79.99 98.58 |
99.9 99.5 99.9 99.8 99.9 98.6 |
Ref [10] | |||||
| Mean ± SD n Variance %SE t-test F-test |
99.36 ± 0.6 6 0.36 0.24 0.768(2.228)* 1.44(5.05)* |
99.60 ± 0.5 6 0.25 0.20 |
||||||||||
| NLT®Vivitrol 50-280 µg mL-1 |
50 100 150 200 240 280 |
49.95 99.14 149.69 200.00 239.36 279.47 |
99.90 99.14 99.79 100.00 99.73 99.81 |
12 16 20 24 28 32 |
11.95 15.89 20.00 23.78 27.94 31.95 |
99.6 99.3 100.0 99.0 99.8 99.8 |
Ref [21] | |||||
| Mean ± SD n Variance %SE t-test F-test |
99.73 ± 0.3 6 0.09 0.12 0.750(2.228)* 1.78(5.05)* |
99.58 ± 0.4 6 0.16 0.16 |
||||||||||
* Figures in parentheses are the tabulated values of t-and F- testes at 95% confidence limit [32]** %SE= SD/
Table 6: Analytical results of the determination of NLB, NLX and NLT in dosage forms using CZE method and reference methods.
| Samples | Ratio of NLB:NLX:NLT |
Taken μg mL-1 |
NLB | NLX | NLT | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Found μg mL-1 |
% Recovery | Found μg mL-1 |
% Recovery |
Found μg mL-1 |
% Recovery |
||||||||
| Serum | 1:1:1 1:2:2 1:4:4 1:5:5 1:6:6 |
20:20:20 20:40:40 20:80:80 20:100:100 20:120:120 |
19.85 39.79 78.56 99.50 119.57 |
99.25 99.47 98.20 99.50 99.64 |
19.69 39.82 79.75 99.90 119.62 |
98.46 99.54 99.69 99.90 99.68 |
19.60 39.84 79.49 99.70 119.48 |
98.00 99.59 99.36 99.70 99.57 |
|||||
| Mean ± SD n Variance **%SE |
99.21 ± 0.6 5 0.36 0.27 |
99.45 ± 0.6 5 0.36 0.27 |
99.24 ± 0.7 5 0.49 0.31 |
||||||||||
| Urine | 1:1:1 1:2:2 1:4:4 1:5:5 1:6:6 |
20:20:20 20:40:40 20:80:80 20:100:100 20:120:120 |
19.69 39.94 79.50 99.33 119.58 |
98.48 99.87 99.38 99.33 99.65 |
19.60 39.65 79.32 99.50 119.28 |
98.00 99.12 99.15 99.50 99.40 |
20.00 39.74 79.50 99.79 118.79 |
100.00 99.35 99.38 99.79 98.99 |
|||||
| Mean ± SD n Variance **%SE |
99.33 ± 0.5 5 0.25 0.22 |
99.03 ± 0.6 5 0.36 0.27 |
99.50 ± 0.4 5 0.16 0.18 |
||||||||||
**%SE= SD/
Table 7: Analytical results of the determination of NLB, NLX and NLT in human serum and urine using CZE method.
The present study introduced a new electrophoretic method for simultaneous separation and determination of opioid agonist nalbuphine hydrochloride and its related antagonists naloxone hydrochloride and naltrexone hydrochloride. The developed method was employed for determination of the selected drugs in their pharmaceutical preparations and biological fluids. Under optimum conditions the proposed method gave excellent separations and detection of the investigated drugs and the obtained results were acceptable with respect to migration time, resolution, and peak area. The described method was also employed for determination of the drugs of interest in human serum and urine, excellent results were obtained. The method was very sensitive, simple, less time consumed and highly precise. Method validation was made to ensure the suitability of the developed method for detection of nalbuphine hydrochloride, naloxone hydrochloride and naltrexone hydrochloride using CZE.
This project was supported by King Saud University, Deanship of Scientific Research College of Science Research Center.