Journal of Clinical Trials
Open Access
ISSN: 2167-0870
ISSN: 2167-0870
Research Article - (2016) Volume 6, Issue 5
Aims: The Treatment in Morning versus Evening (TIME) pilot study sought to establish the feasibility of an onlineonly study detecting whether evening dosing of antihypertensives is more cardio protective than morning dosing. Methods: The TIME study uses a prospective, randomised, open-label, blinded end-point (PROBE) design. In response to various forms of advertising, patients from primary and secondary care, and databases of patients who had previously consented to being contacted about research in the UK, enrolled on the study website (www.timestudy.co.uk). Furthermore, 1,794 hypertensive subjects were written to in three primary care practices as a form of targeted advertising. Participants had to be over 18, prescribed at least one hypertensive drug and have a valid email address. Subjects self-registered, consented, and entered demographics and drug treatments online, before being randomised to taking their antihypertensive therapy in the morning or evening. Automated email followup was used to track patient reported cardiovascular outcomes for the year-long pilot study. Result: 355 participants were randomised and followed up for ≥ 12 months. During this period, 14 participants withdrew from the randomised time of treatment. 59 patients were randomised from 3 practices which wrote to patients publicising the study, giving a rate of 33 randomised per 1,000 patients written to. The 10-year ASSIGN cardiovascular risk of the randomised participants varied by age; 21% for all ages (n=355), 25% for >55 yrs (n=269), 27% for >60 yrs (n=227) and 30% for >65 yrs (n=150). Based on participant cardiovascular risk during the pilot, a full trial with 80% power to detect a 20% improved outcome of nocturnal dosing would require 631 events to occur. Conclusion: The TIME study pilot achieved recruitment efficiently. Based on the pilot data, the TIME study appears viable and has been funded by the British Heart Foundation to recruit over 10,269 subjects and to follow them up for 4 years.
Nocturnal blood pressure (BP) is a better predictor of cardiovascular outcome than daytime blood pressure [1-4]. The Ohasama study demonstrated a linear relationship between reductions in nocturnal blood pressure and reduced cardiovascular mortality [5]. In a study of resistant hypertension, examining the effect of drugs taken in the evening, bedtime dosing was found to be associated with significantly lower 24 hour means of systolic and diastolic BP. This difference was largely driven by greater reductions in asleep BP [6]. The Monitorización Ambulatoria para Predicción de Eventos Cardiovasculares (MAPEC) study results, published in 2010, randomly assigned 2,156 hypertensive clinic patients to taking one or more of their antihypertensive medications at night [7,8]. All patients underwent baseline and annual ambulatory monitoring, and all wore wrist activity meters to control for the effects of activity on BP and to confirm when subjects were asleep [9]. The MAPEC study found that those who took one or more antihypertensive drugs at bed-time had 69 cardiovascular events whereas those who took all their medication in the morning had 187 events, a relative risk reduction of 64% (p<0.001). Unfortunately, given the dramatic results, this study had several limitations in its design and reporting. It is not clear how randomisation was performed. There was no independent adjudication of cardiovascular outcome events and the chosen outcomes events were not easily comparable with other cardiovascular outcome trials.
Further studies are needed to establish the optimal time of dosing for hypertensive patients [10,11]. The Treatment in Morning versus Evening (TIME) study aims to determine whether evening dosing is indeed more effective than morning dosing of antihypertensive at preventing cardiovascular events such as heart attacks and strokes. This paper discusses the feasibility of delivering the formal TIME study, based on pilot data acquired during a 12 month pilot phase.
Purpose
The primary objective of the TIME study is to confirm or refute the conclusions of the MAPEC study by identifying whether nocturnal dosing of antihypertensive medication reduces cardiovascular event rates compared with conventional morning dosing. Secondary questions to be considered ask whether there are any downsides to nocturnal dosing:
Will patients accept nocturnal dosing?
Is nocturnal use of diuretics associated with troublesome nocturia?
Does evening dosing result in nocturnal hypotension, falls or fractures?
There were initial doubts as to the sustainability of the study being conducted online and whether there would be sufficient recruitment Therefore the purpose of the TIME pilot study was to test the feasibility of conducting the full study and to establish the participant recruitment rates. The full trial includes sub-studies of the effects of evening dosing on sleep quality and on cognitive function in older participants. Ethical approval has also been granted to conduct a genetic sub-study using swabs collected from study participants. These were not initiated during the pilot phase. However, a sub-study involving the collecting of BP data provided by participants who own their own BP monitors was started during the pilot phase of the TIME study.
Methodology
The TIME study is a prospective, randomised, open-label, blinded end-point (PROBE design) controlled clinical trial [12]. The TIME pilot is built on successful novel methodology to track patient outcome using Information Technology (IT) [13] and record linkage to identify hospitalisations and deaths within study populations. Endpoints in the full TIME study will be detected more formally using record linkage according to the study protocol [14]. In the pilot phase we used only self-reported events from participants, and surrogate reported events.
The TIME website (www.timestudy.co.uk) is programmed using C#.Net and a Structured Query Language (SQL) Server 2008 database. The website follows best security practices and was designed to be easy to use. All data are captured and managed using the website. Participants self-enrol, consent, and submit follow-up data and home BP readings via the Electronic Case Report Form (eCRF). Follow-up submissions are elicited by automated email. The flow process for the TIME study can be viewed in Figure 1.
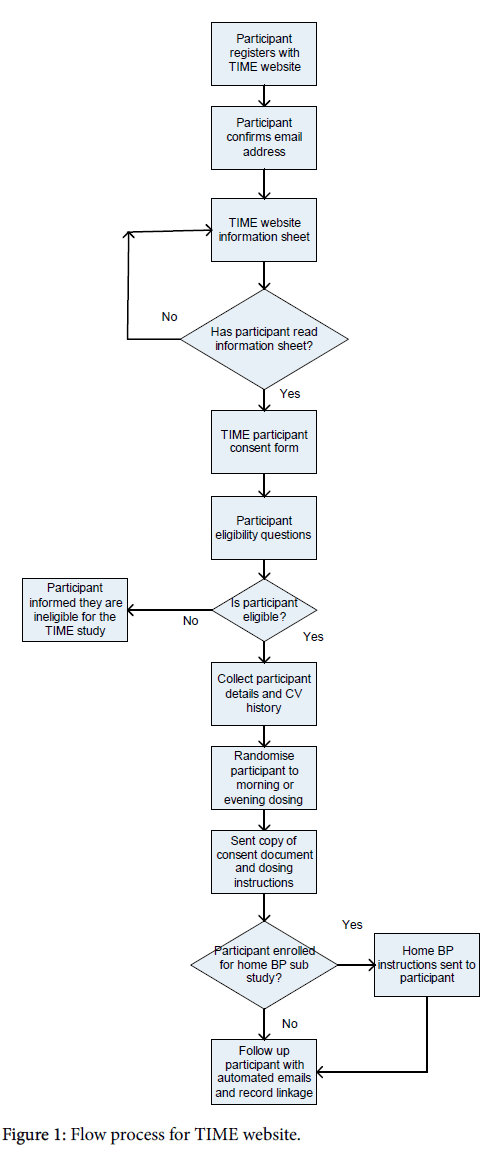
Figure 1: Flow process for TIME website.
Recruitment
Recruitment in the pilot phase was achieved by asking three primary care practices to write to all of their treated hypertensive patients. Patients who signed up were tracked through the eCRF, and data was collected on how participants came to hear about the study. Additionally, posters advertising TIME were sent to all UK GP practices and prescribing cost centres (15,158) with a request to display them in their waiting area [15]..
A YouTube video [16] and social media campaign was launched, with emails sent to all staff in the University of Dundee Medical School. The study was also discussed on Radio 4’s “Inside Health”, in an effort to publicise the study.
The inclusion and exclusion criteria for recruitment are listed in Box 1 and Box 2.

Box 1: TIME Inclusion Criteria.

Box 2: TIME Exclusion Criteria.
Intervention
Randomised subjects were allocated to take their antihypertensive medication at one of two dosing times. Participants allocated to take their BP medication in the morning were asked to take all of their usual BP lowering medications between 6 am and 10 am. Participants allocated to the evening were asked to take all their BP lowering medication between 8 pm and midnight. No other intervention takes place within the study and participants continued to attend their GP or outpatient clinic for routine treatment of their hypertension.
Consent
A patient information sheet was made available on the TIME website with detailed information about the study. Participants were given opportunities to clarify any points they did not understand, and were able to ask for more information by using a 'Contact Us' link, or the Freephone telephone number, on the study website.
Patients were required to fill in an electronic consent form (Figure 2). Electronic consent has been accepted by the National Health Service (NHS) as a viable alternative to a written signature [17]. The consent form consisted of check boxes to attest to consent for each aspect of the trial. A final check box was marked “Checking this box is equivalent to your signature in electronic form. Checking this box means that you agree to participate in the TIME Study”. The consent form recorded the participant’s Internet Protocol (IP) address along with a time stamp of when the form was submitted electronically. A copy of the consent form was sent to participants electronically for their own record. As usual, the decision to participate in this clinical research was voluntary and was based upon the agreement that the potential participant had a clear understanding of what was involved.
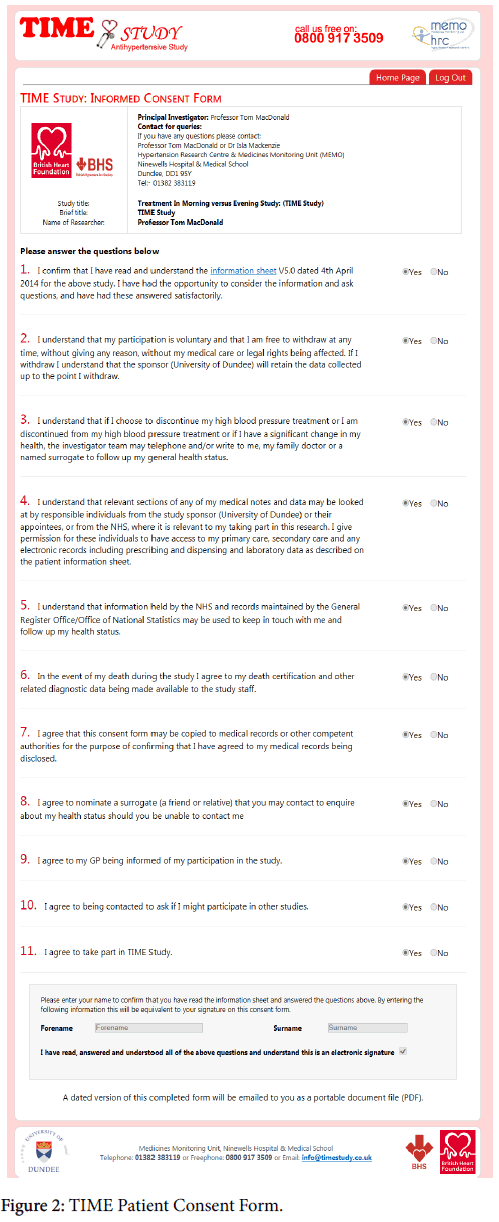
Figure 2: TIME Patient Consent Form.
Patient surrogates
During the process of enrolment, consented participants were asked to nominate a next of kin or surrogate who would be contacted during the trial if the participant failed to respond to follow-up email communications.
Patient reported outcomes
Patients who were randomised into the study submitted patient reported outcomes 1 month after allocation and then every 3 months for the duration of the pilot. Automated emails were used regularly to request “follow-up” data (Figure 3).
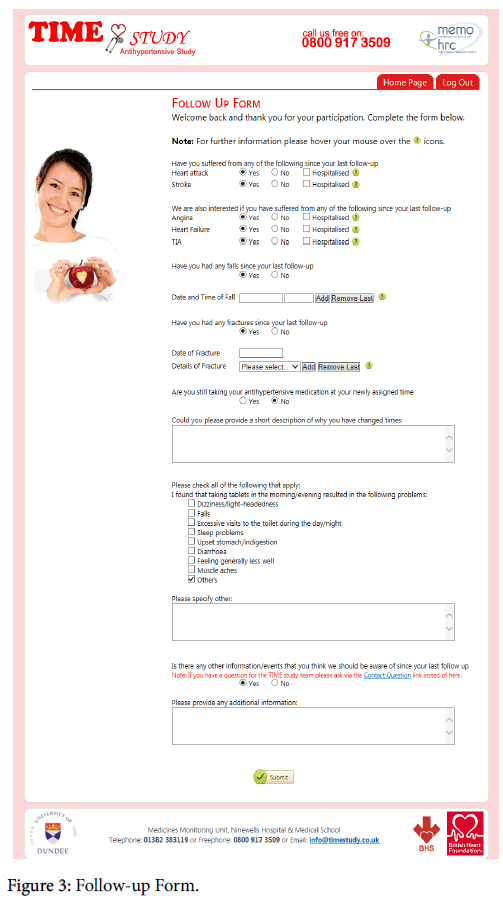
Figure 3: Follow-up Form.
Timely reminders were sent to the participant asking them to complete the online questionnaire. A participant’s nominated surrogate was contacted if a follow-up questionnaire had not been completed within 14 days of a second email reminder.
Patients were not restricted to waiting for automated emails to report changes to their health. Questionnaires could be completed online at any time by logging into the study website.
Withdrawal
Participants were free to withdraw from the study at any point and for any reason. During the pilot, participants could withdraw from: taking their BP medication at their allocated time, receiving further automated emails, or from follow-up by record linkage (including contacting their nominated surrogate or their general practitioner). In order to assess the necessary recruitment for the main study, the pilot study collected data on the number of withdrawals and any reasons given.
Treatment allocation and randomisation
Subjects were randomly allocated to taking all of their blood pressure medications in the morning (6 am to 10 am) or in the evening (8 pm to midnight). Randomisation was achieved using randomly generated bits (0s and 1s. 0 = morning and 1 = evening), which were allocated sequentially to participants as they completed enrolment. The time of allocation was confirmed to participants via automated email, sent within 24 hours.
Study population
The TIME study was open to hypertensive individuals aged 18 or older, in the UK, prescribed one or more once daily antihypertensive drug, and with a valid email address (Box 1).
Blood pressure sub-study
During study enrolment, participants were asked if they owned their own home BP monitor and if they were willing to submit home BP readings. Subjects were instructed to record home BP readings for a week prior to commencing their allocated time of dosing, and then quarterly throughout the pilot study. The BP data was not analysed during the pilot but it is hoped that it will demonstrate any effects of morning versus evening dosing and may help validate adherence to nocturnal dosing during the formal study.
Security and data protection
In keeping with the need for strict maintenance of the security of the electronic case report form (eCRF) [18], the website used the secure hypertext transfer protocol (https). The Server where patient data was held sits behind the University of Dundee firewall and the Medicines Monitoring Unit (MEMO) firewall, located within Ninewells Hospital Dundee. Both firewalls are maintained with regular security updates and are regularly audited. All participant passwords in the database were encrypted and all login attempts to the system were captured and stored along with the IP address and the date of login attempt. If a specified number of unsuccessful login attempts occurred within a specified timeframe the software automatically blocked the IP address from further login attempts. MEMO understands that the weakest point of the login system is the user password and insisted on a minimum password length of 8 characters and required it to contain numeric and alphanumeric characters.
All changes to the trial database were logged allowing the database to be reset to any specific point in time. A record of any individual who made changes was maintained to comply with audit requirements. The software logged all exceptions and errors in the trial database. Errors were automatically emailed directly to study staff ensuring all errors were identified. The pilot study and the main study comply with the requirements of the Data Protection Act 1998 with regard to the collection, storage, processing and disclosure of personal information and uphold the Acts core principles.
Adverse events
The TIME study pilot collected reports of adverse events (AEs) associated with changing the time of dosing. These data were collected during follow-up and at the time of withdrawal from the study. Participants self-reported adverse events thought to be related to the time of dosing. All time of dosing-related reported AEs were recorded in the eCRF. Where there was an AE, participants themselves judged whether they were happy to continue taking treatment at the time randomised or whether they wished to revert to their original time of dosing.
End-points and adjudication
Although independent adjudication of potential study endpoints will be performed in the full TIME study, this was not done in the pilot study. During the pilot there was no formal mechanism for adjudication of patient reported end points. End points reported during the pilot will be adjudicated in the formal study. For full details of the formal study endpoints and adjudication see the TIME protocol [14].
694 individuals registered with the TIME study website during the first year of the pilot. Of those who registered 391 filled in the online consent form and 387 agreed to consent. 355 completed the enrolment process and were randomised. There were 903 follow-ups completed and 499 home BP readings submitted electronically in the first year. 59 of the participants were randomised from the three practices in which hypertensive patients were written to, giving a rate of 33 randomised per 1,000 patients written to. Recruitment from poster advertising was modest, with only 5% of practices displaying the TIME study poster [15]. The social media campaign was not successful and achieved poor recruitment levels, resulting in 4 randomised participants.
The mean age at randomisation within this pilot study was 61 years (median 63). The cardiovascular risk profile of those who enrolled (based on ASSIGN risk score [19]) showed a 10-year risk of 21% averaged over all age groups. Patient retention was good. Over the first-year only 14 subjects withdrew from the randomised time of dosing but 11 of these agreed to email and record-linkage follow-up, while the remainder agreed to record-linkage follow-up (no-one withdrew active consent for follow-up). Fifty-seven subjects did not respond to email contact at some point during the first year but, of these, 24 patient’s nominated surrogate answered on their behalf. 1 participant died; there were 4 reported cerebrovascular accidents and 4 myocardial infarctions.
Pilot data used to power the formal trial
Ten-year predicted CV risk of participants who were randomised into the pilot varied greatly with age, being 21% for all ages (n=355), 25% for those aged >55 years (n=269), 27% for those aged >60 years (n=227) and 30% for those aged >65 years (n=150). Given the age and risk distribution a trial with 80% power to detect a 20% improved outcome of nocturnal dosing will require 631 events to occur. This will require 9,780 subjects of all ages, 8,260 of those aged ≥ 55,7,680 of those aged ≥ 60 or 6,454 of those aged ≥ 65 all followed for 4 years. Figures will be inflated by 5% to accommodate for drop-outs. Recruitment of about 20 subjects per average sized practice (n=5,581) is projected, so TIME needs to recruit approximately 500 practices, if all hypertensive patients are invited. As there is modest cost involved in recruitment, participants of all ages will be recruited.
Home blood pressure results
Two hundred and seven (58%) participants agreed to submit home BP measurements as part of a sub study for TIME. In the pilot study they made 499 BP submissions electronically over 12 months. Feedback on the ease of entering home BP measurements has been positive and there have been no withdrawals from home BP readings. Feedback has led to improvements being made to the website and participants are now able to enter measurements daily as opposed to having to submit a weeks’ worth of measurements at once.
The TIME study requires that subjects have a valid email address and that they are technologically literate in order to self-enrol. This IT literacy requirement may limit the generalizability of the study findings. However, the mean age at enrolment of 61 years appears to be reasonably representative of the hypertensive population. Running a clinical trial online, where participants are entrusted to submit accurate data does raise data accuracy issues [20-22]. However, TIME does corroborate participant-reported data by record linkage of endpoints. Further analysis will be needed to assess the impact of running clinical trials online and to determine how best to ensure accurate data collection.
Quality of communication is critical to optimised healthcare and has been linked with adherence to treatments and patient retention in trials [23,24]. Regular automated emails sent to participants should facilitate retention, adherence, and communication with participants. Ease of use of eCRFs and recruitment processes play a crucial role in improving clinical trial results [25,26]. Validation of the TIME website is important and will involve the use an online questionnaire to gauge participant satisfaction and establish ease of use. Questionnaires have proved a satisfactory method of analysing previous clinical trials [27,28]. In an assessment of the online methodology, an evaluation of cost-per-patient-recruited, retention rates and data entry error rates will be conducted comparing the full TIME study with conventional, paper-based, clinical trials.
The TIME study pilot has randomised 355 participants, with retention being comparable to paper-based trials (96% retention) [22]. During the pilot phase of the TIME study, recruitment suffered from a low response rate to the study invites, similar to conventional paper based trials, but it is hoped that the convenience of taking part in the trial online will prove attractive to potential participants.
The technical objective of the TIME pilot was to produce a low cost, secure IT solution. The pilot study allowed participants to register, consent, enrol and be randomised, whilst adhering to data protection requirements and ethical principles. On the strength of these pilot data, the TIME study rolling pilot has now received a British Heart Foundation grant to write to 300,000 hypertensive patients inviting them to self-enrol. It is hoped that lessons learnt in the TIME Study will enable further clinical trials to adopt similar online methodologies.
If the full TIME study shows definite benefits of dosing antihypertensive medication in the evening rather than the morning, this would represent the most cost-effective advance in the treatment of hypertension and the prevention of CV disease in recent years.
Ethical approval has been obtained from the Tayside Committee on Medical Research Ethics. MREC reference: 11/AL/0309. UKCRN ID: 17071. TIME is registered as ISRCTN: 18157641. The trial is performed in line with Good Clinical Practice guidelines and International Society of Pharmacoepidemiology (ISPE) Good Pharmacoepidemiology Practice Guidance [29].
The idea was conceived by Thomas MacDonald. The study was developed further by the British Hypertension Society (BHS) Research Network working party (Chair Christian Delles (Glasgow)). The initial draft of the present manuscript was created by David Rorie and Thomas MacDonald and circulated amongst the authors for critical revision. All authors approved the final version of the manuscript.
The TIME pilot study was funded by the British Hypertension Society. The full study has been funded by a grant from the British Heart Foundation, Greater London House, 180 Hampstead Road, London, NW1 7AW.