Journal of Osteoporosis and Physical Activity
Open Access
ISSN: 2329-9509
ISSN: 2329-9509
Review Article - (2015) Volume 3, Issue 3
Bone is in a continual state of flux, with old bone being continually replaced by new. This bone turnover, or remodelling, needs to be highly regulated to prevent disorders associated with aberrant bone mass. This process is controlled principally by two cell types, osteoclasts and osteoblasts, which are responsible for bone resorption and deposition respectively. A crucial, well established regulatory mechanism involved in the control of these cells is the RANKL/RANK/ OPG pathway. With this, an osteoblast derived ligand, RANKL, binds to an osteoclastic receptor RANK, producing increased osteoclastogenesis and resorption. In contrast the Ucn system has only recently been found in bone cells, where an osteoblast and osteoclast derived ligand Ucn1, binds to an osteoclast derived receptor CRF-R2β, resulting in inhibition of osteoclastogenesis and resorption. Both systems possess a representative osteoblast derived binding factor with the potential to terminate the ligand signal. In this review we will briefly describe the discovery of the two systems and then go on to compare and contrast the respective components of these two bone regulatory mechanisms. We will review the pathways employed to produce their bone metabolising effects, and finally, we will speculate upon new areas of research which could be exploited to alleviate conditions associated with abnormal bone mass.
<Keywords: Urocortin; RANK/RANKL; Osteoporosis; Osteopetrosis
Ucn 1: Urocortin; CRF-R2β: Corticotropin releasing factor receptor 2 beta; CRF-BP: Corticotropin releasing factor-binding protein; RANK: Receptor activator of NFkB; RANKL: Receptor activator of NFkB ligand; OPG: osteoprotegerin TNF; Tumour necrosis factor; Oc: Osteoclast; Ob: Osteoblast.
Bone is a highly dynamic organ having several disparate physiological functions including: a scaffold for vital organs and muscles, a repository for biologically active cytokines and a reservoir for key ions such as calcium and phosphorus. Its organic component, or osteoid is the non-mineralised extracellular matrix, composed of proteoglycans and collagen. This becomes mineralized by the deposition of hydroxyapatite composed of calcium and phosphorus which represents its inorganic fraction. It is this bone mineral which provides the unique rigidity of bone and distinguishes it from other collagenous matrices [1]. The replacement of old bone with new is a continual process, occurring throughout life and It is estimated that complete skeletal turnover ie the complete replacement of old bone with new, takes approximately 10-25 years in humans [2]. This remodelling of bone (replacing or renewing) is a highly regulated process. However, when imbalances occur in this process, important skeletal diseases result such as osteopetrosis, sclerosteosis and Van Buchem disease (high bone mass), or osteopenia and more severely osteoporosis (reduced bone mass) [3]. The key cellular players responsible for bone turnover and remodelling are the osteoclasts (Ocs) which resorb bone and the osteoblasts (Obs) which lay down new bone. Ocs are large multinucleated cells which are unique in their ability to resorb mineralised bone matrix. They originate from the fusion of cells derived from the monocyte/macrophage lineage of hemopoietic origin. Mature Ocs resorb bone by creating a “ruffled border” composed of podosomes, a complex structure of deeply interfolded finger-like projections of the plasma, and cytoplasmic membranes adjacent to the bone surface. This results in a tight sealing zone, and the reorganisation of actin into ring-like structures [4]. These anatomical rearrangements or polarisations are the prerequisites to resorbtion initiation. Ocs generate these domains only when they are in contact with mineralised matrix. The degradation of the mineral component of bone is brought about by the creation of an acid microenvironment produced by the transport of hydrogen and chloride ions from the ruffled border to the bone surface [5]. As well as creating a highly acidic environment for mineral degradation, the low pH also provides the correct environment for the optimal activity of the protease cathepsin K. This is a key, highly specialised protease which is responsible for the matrix degradation [6]. Unlike Ocs, Obs are derived from mesenchymal stem/progenitor cells. Commitment to the Ob lineage is marked by the temporal expression of the transcription factors Runx2, followed by osterix, both essential for osteoblastogenesis. Ob progenitors differentiate into Obs and express progressively mature markers such as alkaline phosphatase and type I collagen, ultimately becoming terminally differentiated Obs located on the bone surface and expressing markers such as osteocalcin [7]. Obs produce bone by synthesis and directional secretion of type I collagen, which is responsible for over 90% of bone matrix protein. Mineralisation is achieved by the local release of phosphate, which is generated by phosphatases present in Ob-derived, membrane bound matrix vesicles within the osteoid. Together with the abundant calcium in the extracellular fluid, this results in the growth of hydroxyapatite crystals [8].
Discovery of the RANKL/RANK/OPG - Ucn1/CRF-R/CRFBP axes
In order to maintain a correct and healthy bone mass, there must be some interaction/crosstalk between bone cells responsible for mineral deposit and those responsible for resorption. In 1981 the regulation of Ocs by the Ob lineage was still only a hypothesis, but it was strengthened by experiments which showed convincingly that for complete osteoclastogenesis and resorption, a mechanism was needed whereby contaminating Obs that made contact with the precursors of Ocs, was an absolute requirement [9,10]. In 1997, osteoprotegerin (OPG) was identified and its gene encoded a member of the tumour necrosis factor (TNF) receptor family [11]. A year later, the discovery of a related ligand, receptor activator of NFκB ligand (RANKL) was reported and was found to be able to bind both OPG, (its pseudoreceptor) and its true receptor, receptor activator of NFκB (RANK). These discoveries represented a triad of self-regulating molecules which were in effect master controllers of Oc function. RANKL is synthesized in membranous or soluble form by the osteoblastic lineage cells, immune cells, and some cancer cells. This ligand binds to the Oc surface receptor, RANK, and stimulates bone resorption through osteoclastogenesis and the activation of multinucleated mature Ocs. Ob derived OPG, was determined to represent a decoy receptor for RANKL, preventing RANKL from binding to RANK and therefore is an antagonist to both osteoclastogenesis and bone resorption [12,13].
The first member of the Urocortin (Ucn) system, the ligand Ucn1 was isolated in 1995 by its cross-reactivity to antisera against suckerfish urotensin I, a fish peptide structurally related to corticotropin releasing factor (CRF). The first mammalian isoform was cloned from a cDNA library constructed from a portion of the rat midbrain that included the Edinger-Westphal (EW) nucleus [14], and was later found to be expressed in humans [15]. Subsequently, two additional paralogues of Ucn1 were identified Ucn II and Ucn III , having human homologues termed stresscopin related peptide (SRP) and stresscopin (SCP) respectively [16]. These brain related molecules were originally associated with the hypothalamic/pituitary/adrenal axis (HPA) axis and are involved in the stress response in mammals. Specific binding sites for these ligands were also identified as the receptors CRF-R1 and CRF-R2, along with a binding factor to Ucn1 itself, corticotropin releasing factor-binding protein (CRF-BP) [17]. Although originally identified in brain, the Ucn system has now been identified in numerous peripheral tissues where it has effects including: cell proliferation, cell differentiation, cell movement, and cell survival [18-20]. However, unlike the RANKL system, it was not until recently that Ucn1 was demonstrated to be associated with skeletal function [21-23] and even more recently, its expression was confirmed in bone cells along with a CRF receptor subtype and an Ob derived binding protein to Ucn1. Importantly however, unlike RANKL, Ucn1 has opposing effects on bone cells causing both inhibition of osteoclastogenesis and resorption [24]. It is tempting to speculate that similar to the effect of OPG on RANKL, the CRF-BP has the potential to antagonise the effects of Ucn1 in bone and therefore remove the block to osteoclastogenesis and bone resorption. Therefore, making the Ucn system a complimentary self-regulatory system working in an antagonistic manner to that of the RANKL system. It is clear that there are staggering parallels between these two systems, yet functionally they are in opposition. One representing a stimulatory effect and the other inhibitory but ultimately both possessing the potential to regulate bone density.
Next we will look in more detail at the individual protagonists which make up these disparate systems. From this it will become clear that the components of the two systems are vastly different, yet their commonality is to control bone resorption and therefore bone mass.
RANKL/RANK/OPG - Ucn1/CRF-R1/CRF-BP system components
The ligands: RANKL/Ucn1
RANKL belongs to the tumour necrosis factor (TNF) cytokine family and is a type II homotrimeric transmembrane protein. RANKL is composed of 314 amino acids and can exist as 3 isoforms: RANKL 1, 2 and 3. As well as its high expression levels in bone, it is expressed in lung, bone marrow and lymphoid tissues. Typically it is expressed in a membrane-bound form in Obs, but can also become soluble after a proteolytic cleavage by matrix metalloproteases (MMP3 or 7) or a disintegrin and metallopeptidase. However, the membrane bound form is by far the most potent at initiating osteoclastogenesis [25] The importance of RANKL/RANK in bone physiology was made clear when loss-of-function mutations of RANKL and RANK resulted in the high bone mass disease osteopetrosis, this is because Oc
formation was completely impaired. This provided definitive proof for the pivotal role of RANK and RANKL in bone physiology [26,27]. Furthermore, analysis of the RANKL promoter revealed the presence of binding sites for several endogenous molecules known to be involved in bone homeostasis including; vitamin D, and glucocorticoids, and several cytokines including IL1, IL11,IL17, all of which have the ability to regulate RANKL expression [28].
In contrast to the relatively large size of RANKL, the Ucn1 gene encodes a 122 residue pre-peptide, with mature Ucn1 contained within the carboxyl terminus. Ucn 2 and Ucn 3 also exist as prohormones and all three are cleaved by members of the prohormone convertase family into very short active peptides of 40, 38 and 39 amino acids respectively. Each peptide has a unique anatomical distribution and consequently, despite a structural family resemblance, the natural functions of the three Urocortins may differ significantly. Ucn1 is substantially distributed in the periphery [29-31] and thus far it is the only ligand reported to be expressed in Ocs and Ob progenitors [32]. Therefore, Ucn1 in bone may function in an autocrine, or paracrine manner (Figures 1 and 2) to produce its inhibitory effects on osteoclastogenesis and bone resorption. In contrast, Ucn1 cannot affect Ob function directly, as to date, a CRF receptor has only been found to be expressed by Ocs. Several possible transcription-factor binding sites have been identified by sequence homology in the Ucn1 promoter, but few have yet been tested for actually controlling Ucn1 transciptional events. Putative regulatory elements include a TATA box, GATA binding sites, a CCAAT enhancer binding protein (C/EBP) transcription factor binding site, a binding site for the POU domain transcription factor Brn-2, a cyclic adenosine monophosphate (cAMP) responsive element (CRE) and an oestrogen response element. Very interestingly, four base pairs upstream of the CRE site, the Ucn1 promoter contains a consensus half-site for glucocorticoid response elements (GRE), consistent with the ability of glucocorticoids including oestrogen to regulate Ucn1 transcription [33].
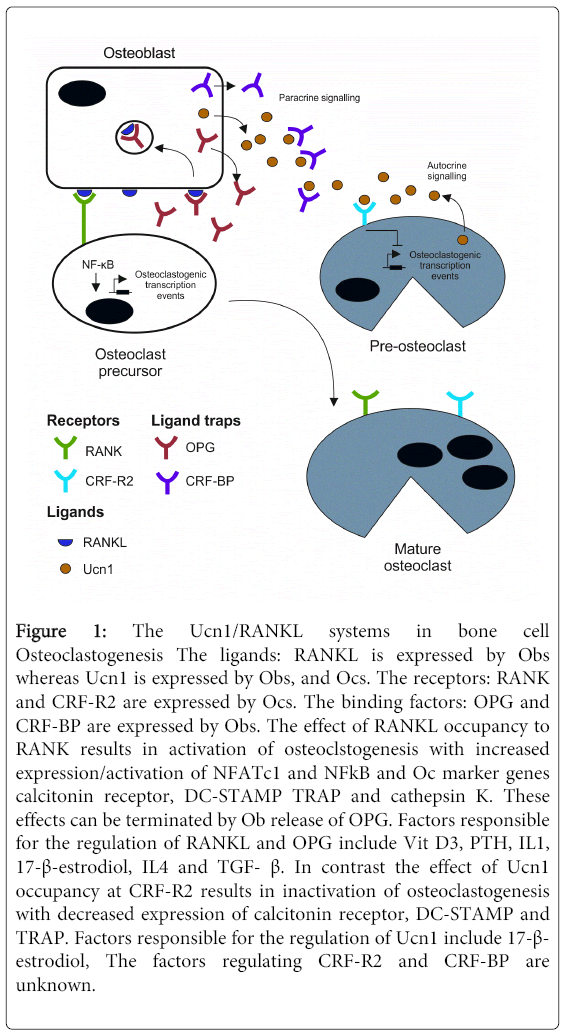
Figure 1: The Ucn1/RANKL systems in bone cell Osteoclastogenesis The ligands: RANKL is expressed by Obs whereas Ucn1 is expressed by Obs, and Ocs. The receptors: RANK and CRF-R2 are expressed by Ocs. The binding factors: OPG and CRF-BP are expressed by Obs. The effect of RANKL occupancy to RANK results in activation of osteoclstogenesis with increased expression/activation of NFATc1 and NFkB and Oc marker genes calcitonin receptor, DC-STAMP TRAP and cathepsin K. These effects can be terminated by Ob release of OPG. Factors responsible for the regulation of RANKL and OPG include Vit D3, PTH, IL1, 17-β-estrodiol, IL4 and TGF- β. In contrast the effect of Ucn1 occupancy at CRF-R2 results in inactivation of osteoclastogenesis with decreased expression of calcitonin receptor, DC-STAMP and TRAP. Factors responsible for the regulation of Ucn1 include 17-β- estrodiol, The factors regulating CRF-R2 and CRF-BP are unknown.
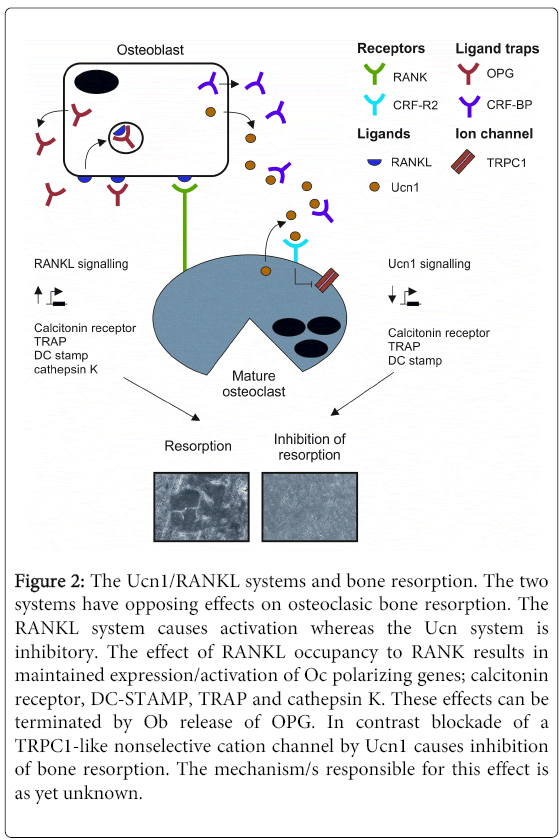
Figure 2: The Ucn1/RANKL systems and bone resorption. The two systems have opposing effects on osteoclasic bone resorption. The RANKL system causes activation whereas the Ucn system is inhibitory. The effect of RANKL occupancy to RANK results in maintained expression/activation of Oc polarizing genes; calcitonin receptor, DC-STAMP, TRAP and cathepsin K. These effects can be terminated by Ob release of OPG. In contrast blockade of a TRPC1-like nonselective cation channel by Ucn1 causes inhibition of bone resorption. The mechanism/s responsible for this effect is as yet unknown.
The receptors: RANK/CRF-R2β
RANK belongs to the TNF receptor superfamily of transmembrane proteins and is synthesized as a type I homotrimeric transmembrane protein. It can be broken down into an extracellular domain consisting of 84 amino acids, a hydrophobic transmembrane domain consisting of 21 amino acids, and a large cytoplasmic domain of 383 amino acids. It is expressed by various tissues such as skeletal muscle, thymus, liver, colon, mammary glands, prostate, pancreas, and cells of the monocyte/ macrophage lineage including Ocs.
In bone, Rank is the specific receptor for RANKL and is expressed on the surface of Oc precursors and mature Ocs. Upon binding of RANKL, RANK activates numerous signalling molecules and transcription factors depending on whether RANK activation occurs in pre Ocs or mature fused multinuclear cells with the capacity to initiate resorption [34]. After RANKL binding, RANK recruits tumour necrosis factor receptor associated factor 6 (TRAF6) to its cytoplasmic domain. This induces NF-kB activation which translocate to the nucleus. At the nucleus NF-kB elevates c-Fos, and AP-1expression which interact with nuclear factor of activated T-cells cytoplasmic 1 (NFATc1) to trigger osteoclastogenic gene transcriptional events. Using mouse models, it was determined that NFATc1represents the master controller of osteoclastogenesis [35,36]. Downstream transcription factors regulate expression of essential Oc genes, such as dendritic cell-specific transmembrane protein (DC-STAMP), tartrate resistant acid phosphatase (TRAP), cathepsin K, matrix metalloproteinase 9 (MMP-9) and calcitonin receptor, which allow the final differentiation and fusion of the precursors culminating in the large, fused multinucleated TRAP positive mature Ocs with the potential to resorb bone. Once they have been produced, the polarisation and initiation of mature Oc resorption involves the rearrangement of the actin cytoskeleton to form F-actin rings creating the characteristic podosome structures [37]. At the bone Oc interface, the αvβ3-integrin (the vitronectin receptor) mediates attachment through the formation of a signalling complex consisting of the tyrosine kinases c-Src, and activation of small GTPases RAC and Rho [38,39]. These enzymes require post-translational modification by isoprenylation to localise correctly in the cell and to exert their specific functions. Unlike the RANK, there are two predominant Ucn1 binding sites, CRF-R1 and CRF-R2, both products of independent genes and encode members of the class B1 (“secretin-like”) receptor family. These are members of the extended G-protein coupled, seven transmembrane spanning domain receptor superfamily. CRF-R2 and CRF-R2 share high sequence homology (70% at the amino acid level) differing predominantly at the N-terminus, and have unique tissue distributions and pharmacological affinity profiles, implying a diversity of function [40-42]. Multiple splice variant isoforms have been observed for each receptor subtype, and include both membranebound and soluble variants [43,44]. At least eight alternatively spliced transcripts of CRF-R1 have been identified in humans, three in rats, four in mice, and nine in hamsters. However, only one of these splice variants is known to induce signal transduction in humans and rodents CRF-R1α [45,46]. Four major CRF-R2 splice variants have been identified, including membrane-bound and soluble isoforms of CRF-R2α, and membrane-bound CRF-R2β and CRF-R2γ receptors [47,48]. Membrane-bound CRF-R2 receptors are respectively 411, 431 and 397 residues in length, [49,50]. Although they have a similar affinity for Ucn 1 as the CRF-R1 receptors, unlike CRF-R1 they also bind exclusively to Ucn II and Ucn III ligands, [51,42]. To date however, in bone, only expression of the CRF-R2β isoform has been detected and this has been demonstrated to be localised exclusively to pre- and mature Ocs [24]. The downstream signaling pathways of CRF receptors is typically diverse. Although they are G-protein coupled receptors, they are able to couple to different G-proteins, principally Gαs and Gαq. The consequence of this is the regulation of diverse intracellular networks activated through adenylate cyclase, or phospholipase C, therefore generating cAMP, or IP3 and diacylglycerol [52-54]. Downstream of these events, numerous effector systems can be activated including intracellular ions, an array of protein kinases, ion channels, and altered gene expression [55,56]. Common signalling pathways utilised by these receptors include: mitogen-activated protein kinase (MAPK) pathways, in particular the extracellular signal-regulated kinases (ERKs). Although the downstream effects of Ucn1 binding to its receptor have been well documented in other tissues, in the cells of the bone there is very little information regarding its role in Oc differentiation, inhibition of resorption. Activation of CRF-R2β by Ucn1 in Ocs resulted in significant inhibition of the formation of fused multinucleated mature Ocs. Presumably by the inhibition of genes related to cell fusion, DCStamp and those responsible for Oc polarization and activation, the calcitonin receptor, TRAP and Cathepsin K [24]. Although not yet observed in Ocs, only Ucn2 has the ability to inactivate NFATc1 in cardiac myocytes and inhibit NFkB transcriptional events [57]. However in rat PC12 cells, Ucn1 was able to activate NFATc1 [58]. Currently there is no data on the effect of Ucn1 on the expression/ activity of NFATc1 or NFkB in bone cells. As well as directly antagonizing some of the genes that are activated by RANK, Ucn1 also uniquely abrogated mature Oc resorptive activity by inhibiting a TRPC1 like cation channel which caused both the inhibition of resorption pit excavations and actin ring generation [24]. These channels are involved in calcium mobilization from extracellular and intracellular sources and are becoming important signalling molecules within their own right. TRPC channels are intimately associated with the regulation of Ca++ concentrations within many cells and in particular, when associated with ORAI and STIM proteins. This produces functional store operated channels, which tightly control Ca++ levels within intracellular compartments in particular the endoplasmic reticulum. This is particularly important in Oc where Ca+ + handling is crucial for their proper functioning [59]. Furthermore, very recently, the TRPC1 channel has been implicated in the regulation of Oc formation and function [60].
The ligand trap: OPG/ CRF-BP
OPG also belongs to the TNF receptor superfamily, and is a protein of 380 amino acids in length. Its sole purpose is to prevent the biological effects of RANKL. Like RANKL, It has several pseudonyms because of its noted actions in different cell systems, these include; TNFRS member 11B (TNFRS11B), osteoclastogenesis inhibitory factor (OCIF) and tropine reductase 1 (TR1). It is highly expressed as a soluble protein and is closely related to CD40. It is produced in several organ systems including lung, heart, kidney, liver, bone marrow, Obs, vascular smooth muscle cells, B-lymphocytes, and articular chondrocytes [61] After the formation of the OPG/RANKL complex is complete, because RANKL is membrane bound the complex is internalized, this is mediated either through lipid rafts or by the clathrin coat formation pathway. These two mechanisms control the bioavailability of extracellular OPG [62]. The importance of OPG to bone homeostasis was highlighted when over expression of OPG in mice resulted in osteopetrosis and its deficiency caused osteoporosis [63]. OPG expression in Obs is increased by vitamin D3, interleukin (IL)-1α, IL-1β, TNFα, TNFβ, BMP2, transforming growth factor β (TGFβ), 17-estradiol and Wnt signaling pathway. Its expression is decreased by prostaglandin E2 (PGE2), parathyroid hormone (PTH), glucocorticoids and insulin like growth factor- 1 (IGF-1). CRF-BP is an evolutionarily conserved secreted glycoprotein that binds Ucn1 with high affinity [64,65]. Cloned from human liver, rat cerebral cortex and mouse brain, CRF-BP cDNAs encode a protein consisting of approximately 322 residues and with a molecular mass of 37-kDa. Unlike OPG, CRF-BP occurs exclusively as a soluble protein but was found to be smaller than the known receptors for Ucn1. It also lacked the consensus sequences found in the transmembrane spanning domains of typical G protein coupled receptors, or a phosphatidyl inositol anchor signal. These observations therefore, led to the suggestion that its function was not that of a signal transducing receptor but as a ligand trap causing a depletion/inhibition of the agonist signal. CRF-BP is now recognised to dimerizes after binding and clear its ligand from the surrounding milieu. Similarly to OPG, CRF-BP has been hypothesized to limit CRF receptor agonist effects by sequestering secreted ligand and facilitating subsequent enzymatic degradation, thereby limiting Ucn1 bioavailability [66,67]. It is found in several cell systems and can co-localise with Ucn1 positive cells, or it can be expressed in Ucn1 negative cells within the same microenvironment. Recently in bone, it has been found to be highly expressed exclusively in Obs with a complete absence of expression levels in Ocs. This raises the intriguing possibility that like Ob derived OPG, Ob derived CRF-BP could sequester local Ucn1 levels, thus removing Ucn1 derived osteoclasic inhibition. Unlike OPG, in bone nothing is known of the stimuli or mechanisms by which CRF-BP is released from cells. However, In the hypothalamus, it appears to be under positive glucocorticoid control [68].
This is an exciting time in the study of the peripheral effects of the Ucn system and in particular its novel and profound effects on bone physiology. This review highlights the potential importance of the newly discovered Ucn1 system in the control of bone density. Like the well established RANKL system, it too is a triad of molecules, involving a ligand, receptor and a binding protein, making the system potentially self- regulating. Like the RANKL pathway, component parts of this system are differentially expressed in bone cells. However, the effects on bone cells are in direct opposition to those of the RANKL pathway. Research involving the role of the Ucn1 system in bone is in its infancy and several important questions remain to be answered. Importantly, how is the bio-availability and expression of the component parts controlled at the molecular level? As factors which regulate the expression/release of the Ucn1 system components may prove useful in the control of Oc generation and activity. An obvious initial anti-resorptive treatment would be the administration of Ucn1 itself, or the development of CRF-R2 receptor specific synthetic agonists. However, this is untenable because of the widespread distribution of both Ucn1 and its receptor throughout the body. Furthermore, when applied systemically, detrimental effects of Ucn1 have been observed in the past, including effects on heart rate and blood pressure. Alternatively, the generation of a fully humanised antibody to CRF-BP, analogous to denusimab which targets RANKL, may be of some use for conditions presenting with aberrant bone mass. Although a specific ion channel, TRPC1 appears to be involved in the anti-resorptive effect of Ucn1, the mechanisms involved are unknown. However, if the use of Ucn1 could be bye-passed and instead, TRPC1 specific openers and inactivators developed, this may represent a novel route for the regulation of bone density. To date, the roles of the other Ucn paralogues, Ucn II and Ucn III have not been investigated in bone. It is known that Ucn II and III are selective for the CRF-R2 receptor subtype, the only receptor found in Ocs. Could these paralogues therefore produce an even greater effect to that already seen with Ucn1? Also of considerable interest is the potential interaction between the RANKL/ Ucn1 systems.
Modulation of such an interaction may allow for the fine tuning of bone remodelling. Unlike the extensive in-vivo studies involving the RANKL system, no such studies have been undertaken involving the Ucn system in bone. There are several genetically modified animal models involving genes deleted for each component of the Ucn system. However, none has focussed on a bone phenotype. Ideally, a conditional Oc specific CRF-R2 KO, or Ucn1 KO animal would be expected to produce an osteoporotic phenotype. In contrast an Ob conditional CRF-BP KO animal would be expected to present with an opposite osteoperotic phenotype. Lastly an interesting and extremely relevant finding is the relationship with Ucn1 and oestrogen. If oestrogen is responsible for the expression levels of Ucn1 in bone ,[69] then the oestrogen/Ucn1 axis may be important in the development of postmenopausal osteoporosis. The relationship can be easily assessed using a Ucn1 KO animal model undergoing ovariectomy (OVX). In WT OVX animals, HRT would be expected to reverse the decrease in bone mass. However, in Ucn1 KO animals, the effects of OVX would not be restored by HRT. Currently there is little information concerning relationships between Ucn1 and factors involved in bone metabolism. However, differential expression of Ucn1 has been found in osteoprogenitor cells by both BMP-2, and TGF-beta-1[32]. Furthermore, although not associated with bone, Ucn1 did cause Increased LPS-Induced Endothelial Permeability by Regulating the Cadherin–Catenin Complex via Corticotrophin-Releasing Hormone Receptor 2, the receptor associated with Oc activity [70] and can modulate GSK3β in cardiomyocytes [71].
It is clear much work remains if we are to fully elucidate the role of the Ucn system in bone physiology and its relationship to the other important regulatory factors. These efforts however, may reward us with several new approaches for the development of novel treatments for bone mass disorders.