Virology & Mycology
Open Access
ISSN: 2161-0517
ISSN: 2161-0517
Research Article - (2023)Volume 12, Issue 4
Cases of the monkeypox virus have been recorded in non-endemic nations and have continued to be reported in several endemic nations since early May 2022. In this study, we modeled Monkeypox Virus (MPXV) Thymidylate Kinase (TMPK) and scaffolding protein (D13) and these models and their templates were taken for small molecule screening against 602,413 small molecules using pharmacophore modeling and molecular docking methods. Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) properties were also computed followed by Molecular simulation dynamics studies. All presented hits had superior molecular docking scores to used reference standards of Cidofovir and Rifampicin. TMPK compounds displayed better ADMET profiles than D13 compounds, hence the latter may necessitate ligand optimization. Following molecular dynamics simulation, analysis revealed that all generated complexes were stable, with the ligands NPC275538, NPC244454, 135566871 and CHEBI compounds outperforming other hits. These compounds still presented higher docking scores against cidofovir-resistant TMPK and Rifampicin-resistant D13 proteins. Compounds 447970, 446595 and 54723327 were most selective against human TMPK. The conserved interaction patterns of these compounds among vaccinia and monkeypox virus proteins with the fact that studied proteins are highly conserved across Orthopoxviruses (OPV) is appealing that these hits should be studied across OPV. Therefore, these compounds should be subjected to laboratory testing to prove their antipox capability. Since there are currently no approved MPXV antivirals, this discovery significantly aids in developing new drugs for treating monkeypox.
Monkeypox virus; Thymidylate kinase; Scaffolding protein; Pharmacophore modeling; ADMET Studies; Molecular simulation dynamics
The Poxviridae family of viruses is subdivided into Chordopoxvirinae (18 genera) and Entomopoxvirinae (5 genera) subfamilies [1]. Orthopoxvirus (OPV) is perhaps the most notorious among the Chordopoxvirinaea subfamily. It includes the horsepox virus, Monkeypox virus, Vaccinia virus and Variola (smallpox) virus among others. They are housed in a dumbbell-shaped nucleoprotein and have some of the largest and most complex genomes (130-360 kb, 200 genes) known for animal viruses [2]. The poxviruses are known to have large, enveloped, slightly pleomorphic oval-shaped virions measuring 200-400 nm ( ̴ 280 by 220 nm), with a wide host range [3]. Due to the recent appearance of human monkeypox and potential bioterrorism concerns stemming from the poxvirus’s clear capacity to spread swiftly among humans, there has been an increase in concerns about it [4].
TMPK catalyzes the phosphorylation of thymidine 5’-monophosphate (dTMP) to form thymidine 5’-diphosphate (dTDP), a reaction that requires ATP and Mg2+, this is afterward transformed to thymidine 5’-triphosphate (dTTP) by a Nucleoside-Diphosphate Kinase (NDK) [5]. Native Medicare Charitable Trust (NMCT), rutaecarpine, Tipranavir and Cefiderocol have been reported to be repurposable against MTMPK while cidofovir and acyclovir are also reported in managing MPXV symptoms [6-10]. Cidofovir-Resistant (CDV-R) MPXV has been reported [11]. Several nucleot(s)ide analogs are available that inhibit OPVs [12].
Rifampicin targets the scaffolding protein, D13 and reversibly impairs crescent membrane formation before the Immature Virus stage and results in the relocation of D13 into cytoplasmic inclusion bodies [13]. Rifampicin prevents the binding of A17 (integral membrane protein necessary for D13 honeycomb lattice formation), occupying the phenylalanine-rich pocket on the 3-fold axis of the D13 homotrimer resulting in the production of an abnormal virion. However, resistance to this antibiotic has been reported [14-17]. OPV D13 orthologs have high levels of sequence conservation and there may be sequence similarities between D13 and the Major Capsid Proteins (MCPs) of some large eukaryotic dsDNA viruses, according to a comparative genomic study [18]. Simeprevir has been suggested to be a D13 inhibitor using in silico studies. Intertrimer interactions that govern the D13 assembly are however been poorly resolved. The goal of this research was to screen for TMPK and D13 inhibitors from MPXV and their templates from VACV since there aren't any antivirals that have been given the green light yet [6].
Structures used in the study
The protein structures for MPXV were obtained by homology modeling while those for Vaccinia Virus (VACV) were downloaded from The Protein Data Bank (PDB), TMPK (PB ID: 2V54) and D13 (PDB ID: 6BEB) [19]. 602,413 Small molecules were screened and downloaded from their respective databases of CHEBI, PubChem, Drug Bank, National Cancer Institute (NCI) and Natural Product Activity and Species Source (NPASS).
Homology modeling
Homology modeling of MPXV proteins was done using the Swiss model web server and VACV proteins as templates. (Protein Sequence Accessions: YP_010377155.1 and YP_010377107.1 for MPXV TMPK and D13 respectively). The obtained models were submitted to PROCHECK at the SAVESv.6 servers for validation using the Ramachandran plot. The TMPK and D13 models were refined at the galaxy and FG-MD webservers respectively [20].
Pharmacophore screening
The multi-ligand pharmacophore hypothesis to screen for TMPK inhibitors was generated using the Schrodinger phase in defaulted settings by finding the best alignment and common features among the ligands [21].
Glide docking
The proteins were initially preprocessed, hydrogen bonding was optimized and energy was minimized using the default settings of the protein preparation wizard. For TMPK, active waters and Mg2+ were left in the prepared monomeric proteins whereas D13 proteins were left in Homo trimeric form. Protein docking centers were generated by the centroid of workspace ligands using Thymidine diphosphate Thymidine diphosphate (TDP) and Rifampicin in TMPK and D13 respectively. Small molecule preparation was done with LigPrep in defaulted mode [22]. The prepared proteins and ligands were subjected to Glide XP docking.
GOLD docking
To carry out docking in GOLD 5.3.0, the TMPK proteins were loaded into the Hermes 1.7.0, hydrogens were added, ligand and active waters were extracted and the latter were configured before docking and all other water molecules were deleted. The binding sites were defined by ligands and the chem score kinase template was used with TDP as a reference ligand. All H-bond donors/ acceptors were treated as solvent accessible. Template configuration was left default, CHEMPLP fitness function was used with both scoring and rescoring with the same parameter file, default GOLD parameters were used with early termination allowed and internal ligand energy offset, slow and automatic GA search with 100% efficiency was used [23,24]. The metal was automatically assigned octahedral coordination.
Binding energy
Binding affinities were determined by Molegro Virtual Docker (MVD) (2013 6.0.1). Briefly, the proteins were imported into the MVD and prepared by repairing amino acid residues. The ligands in MOL format were also imported and auto-prepared. The docking function was set to MolDock Score and set to enforce H-bond directionality with a user-defined Ligand origin and radius of 13 Å with Ligand evaluation including internal ES, Internal H-Bond and sp2-sp2 Torsions. The algorithm used was MolDock Optimizer with 20 runs and set to minimize energy after docking while parameter settings defaulted and set to return a maximum of 50 poses per run and docking was executed in separate processes. The Ligand pose with the highest re-rank for each Ligand was reported rather than that with the highest MolDock Score/Binding Affinity [25].
ADMET studies
ADMET properties of all the prepared compounds were studied using Schrodinger’s QikProp with settings defaulted [26]. Pan- Assay Interference Compounds (PAINS) alerts were checked by the FAFDRUG4 server and only ligands without PAINS alerts were retained. All the calculated properties were compared to Cidofovir (for TMPK) and Rifampicin (for D13).
Molecular dynamics simulations
MDS was carried out with Gromacs version 2021.4 with GROMOS96 43a1 forcefield on 5 complexes random for each protein [27]. Gromacs pdb2 gmx was used to generate the protein topologies, while PRODRG generated the ligand topologies [28]. All simulated systems were neutralized with the required quantity of counterions (Na+ and Cl-). Extended Simple Point Charges (SPC/E) water molecules were used to solvate the neutralized system in a hexahedral box. All MD simulations were run at 300 K in an explicit solvent. Following that, the system were heated to 300 K with positional restrictions (force constant: 50 kcal). Production MD simulations lasted for 50 ns and it was monitored by checking energies during the simulations. VMD, Pymol and Gromacs binaries were used for the analysis of the results [27].
Homology modeling
The MTMPK template had a Global Model Quality Estimation (GMQE) of 0.98, Quaternary Structure Quality Estimation (QSQE) of 0.79, sequence identity of 98.53%, sequence coverage of 100% with only 3 mismatches were T103, I139 and E148 in the target are replaced with A103, V139 and T148 in the template respectively (Figure S1). Its Root Mean Square Deviation (RMSD) aligned with its template was 0.234 Å. The refined model had an RMSD of 0.283 Å from the initial model without poor rotamers and 96.7% Ramachandran favored residues as compared to 92.9% in the initial model (Figure S2).
The VACV D13 template had a GMQE of 0.97, a QSQE of 0.93, sequence identity of 98.73%, 95.66% sequence coverage with first 25 Indels and more 7 substitutions with residues S40, I135, I345, I105, S112, G538, V539 in the template replaced with F, V, V, V, T, D and I in the model respectively (Figure S3). The refined model of MD13 had an RMSD of 0.402 Å from the submitted model and 0.478 Å when superimposed to its template. Its Ramachandran- favored residues increased from 80.0 to 89.3% upon refining (Figure S4). These results show that templates were very reliable, accurate and valid [29,30]. These models were further taken for molecular dynamics studies.
Pharmacophore screening, molecular docking and binding energy
The pharmacophoric results are shown in Figure 1 and Table 1 for the presented TMPK Ligands. All ligands whose Fitness scores were ≥ 1.200 were retained for further studies.

Figure 1: Features of the used pharmacophoric model for TMPK inhibitor screening.
| Structure | CID | Fitness | Align score | Compound data base |
|---|---|---|---|---|
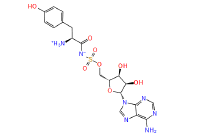 |
449029 | 1.407 | 0.784 | Drug bank |
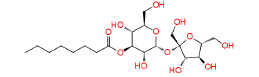 |
448657 | 1.282 | 0.869 | Drug bank |
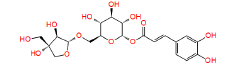 |
NPC473924 | 1.237 | 0.833 | NPASS |
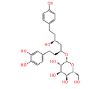 |
NPC79715 | 1.494 | 0.741 | NPASS |
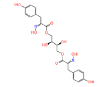 |
NPC275538 | 1.247 | 0.995 | NPASS |
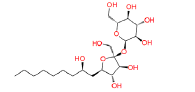 |
17754184 | 1.288 | 0.849 | Drug bank |
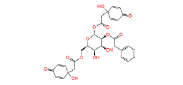 |
NPC244454 | 1.54 | 0.945 | NPASS |
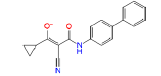 |
54723327 | 1.414 | 0.473 | Drug bank |
 |
447688 | 1.382 | 0.83 | Drug bank |
 |
49866943 | 2.582 | 0 | Drug bank |
 |
446595 | 1.241 | 0.852 | Drug bank |
 |
447970 | 1.644 | 0.488 | Drug bank |
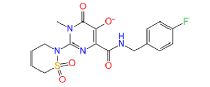 |
54682040 | 1.258 | 0.527 | Drug bank |
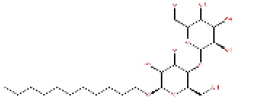 |
447793 | 1.567 | 0.945 | Drug Bank |
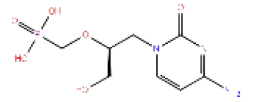 |
Cidofovir | 1.345 | 0.765 | PubChem |
| Note: CID: Compound Identity; NPASS: Natural Product Activity and Species Source; TMPK: Thymidylate Kinase. | ||||
Table 1: The results of pharmacophore (ligand-based) screening for TMPK Inhibitors. The minimum and maximum fitness score and align score values that can be obtained are -1.0 to 3.0 and 0.0 to 1.0 respectively.
The molecular docking results of TMPK were in agreement with pharmacophore study results. The best compound for MTMPK was 17754184 from Drug Bank with XP Glide docking score of -11.293 Kcal/mol, PLP fitness of 85.808, Binding Energy of -185.737 Kcal/mol and H-Bond energy of -21.294 Kcal/mol. The results of molecular docking for vaccinia virus TMPK, Compound 449029 from Drug Bank had the highest XP Glide docking score of -12.300 Kcal/mol, with Binding Energy of -230.343 Kcal/mol, PLP fitness of 112.682 and H-Bond energy of -9.078 Kcal/mol.
The molecular docking results show that these identified hits interact with active site residues involved in the conversion of TMP to TDP and also show highly converging interaction patterns between MPXV and VACV proteins, the TMPK interacting residues are the same for the two viral proteins except ASP50 and THR19 interacting in VTMPK but not MTMPK as well as PHE38 and ASP92 interacting in MTMPK and not in VTMPK [31]. These residues also highly overlap with those identified in Variola virus TMPK (Lys17, Asn37, Asp92, Tyr101, Arg41, Arg93, Asp13, Arg72) except Tyr144 this therefore, suggests that screened compounds may be able to bind other OPV TMPK enzymes with the same mechanism due to their overlapping sequences (Figures S5 and S6) [9,32].
When CDV-R MTMPK was docked with compounds in this study, the docking scores were not significantly different from those of WT-TMPKs (paired t-test p-value of 0.315847 at 95% confidence) and with higher variance compound. 449029 had the highest score of -12.4053, ligands still retained their vital residue interactions with high docking scores, suggesting that identified compounds can still bind CDV-R MPXV and VACV (since CDV-R mechanisms seem to be conserved in both with high Docking scores (and perhaps high Binding Affinities) (Tables S1 and S2) [11]. Due to OPV TMPK similarity (42%) with human TMPK (hTMPK), the compounds presented here were also docked in the active site of Human TMPK to check for their selectivity and the results are presented in, the hTMPK docking scores were significantly lower than those of MTMPK (p=4.97 E-07, at 95% confidence) however hTMPK docking scores had higher variance with 7 Ligands having docking scores ≥ 7.00 Kcalmol-1 and further studies including selectivity optimization and molecular dynamics are needed to characterize the stability of their interactions with hTMPK. Compounds 54723327 and 446595 were more selective against hTMPK (Tables S3 and S4) [31].
The docking scores of VTMPK were significantly higher than those of MTMPK (2-tailed paired t-test value of 0.01967 at 95% confidence) (Table S5). There were no significant differences between PLP Fitness and binding energies of MTMPK and VTMPK (p values of 0.263 and 0.363 respectively at 95% confidence) suggesting the efficiency of these ligands in binding to both viral proteins (Tables S6 and S7).
The best ranking compound for both D13 proteins was CHEBI:2347 with XP Glide docking scores of -10.304 and -9.894 Kcal/mol and Binding Energies of -203.413 and -186.685 Kcal/mol for Monkeypox and vaccinia proteins respectively. D13 docking results also show that most of the interacting residues are the same for MD13 and VD13 but there is a clear-cut difference in which a specific chain is involved in the interaction though this difference may not be significant for Rigid Receptor Docking (RRD) as they are specific to a single top-ranked pose/frame unless validated by MD simulations. The monkeypox D13 specific residues include ASN20 (A), ASN20 (B), LYS469 (B), LYS159 (A), VAL164 (A), VAL164 (B), ASP166 (C) as comparted to PHE486 (A), GLN27 (C), ASP25 (C), SER19 (C), LYS225 (C), LYS17 (B), PHE168(B), LYS225 (A), ASP20 (C) for VACV.
The rifampicin-resistant D13 proteins containing all 11 independent spontaneous mutations were also docked with D13 ligands identified in this study and results show that the obtained docking scores were not significantly different from those of WT- D13 ( p values of 0.260 and 0.176 for MD13 and VD13 respectively at 95% confidence) but with high variance values and Ligands 11986, 11987 and 11988 having reduced docking scores for both targets, Rifampicin scores dropped by over 50% while docking scores for other ligands were relatively constant with CHEBI compounds having highest values and CHEBI:426 having relatively higher scores than for wild type proteins suggesting that these compounds can still bind Rifampin resistant proteins with higher docking scores (Tables S8-S15). It is however crucial that the reader understands that these amino acid substitutions are spontaneous and were not necessarily identified in the same protein minimizing their chances of occurrence in the same protein. It is worth noting that compounds 11986, 11987 and 11988 are bioisosteres and form the same cluster in the lower left corner when grouped according to their ADMET properties (Figure S7) [16]. This, therefore, provides a scaffold on which other compounds can be built while optimizing Hits to inhibit OPV morphogenesis. All Ligands had superior results to used reference standards of Cidofovir and Rifampicin.
ADMET studies
The pharmacodynamics and pharmacokinetics of effective and safe medications are always finely adjusted, with high potency, affinity and selectivity against the molecular target, as well as sufficient absorption, distribution, metabolism, excretion and tolerable toxicity ADMET [33]. The results of ADMET studies are summarized in Figures 2 and 3 with a comparison to Cidofovir and Rifampicin for TMPK and D13 Ligands respectively. The PCA analysis based on all the 51 calculated properties. Most TMPK Ligands formed a cluster in the right middle whereas cidofovir, 54723327, 54682040 and NPC244454 each formed independent clusters (Figure S8). Ligands 11986, 11987 and 11988 formed a cluster in the left bottom corner showing related properties, CHEBI compounds and 135566871 also formed a cluster nearly placed in the center showing closely related properties. Rifampicin, 157232835 and g0mol 79715 also formed independently isolated clusters (Figure S7).

Figure 2: The figure shows how the chosen main ADMET properties for identified TMPK ligands compare to Cidofovir, an FDA-approved acyclic phosphonate analog of deoxynucleoside monophosphates. The Ligand efficiency is (docking score)/(1 + ln (number of heavy atoms).

Figure 3: The figure shows how selected ADMET properties for D13 studied inhibitors compare with FDA-approved Rifampicin. The Ligand efficiency is (docking score)/(1 + ln(number of heavy atoms).
However, it should be emphasized that all the ranges for the ADMET qualities are more intended to serve as guidelines than absolute cutoffs and with regard to Qikprop (For which all properties are referenced in this section), they only apply to oral medications and imply passive transport across a biological system; otherwise, some drugs are not administered orally and others may be absorbed across the cell membrane by active transport, for example, those drugs that are structurally similar to endogenous substances [26,34,35].
All compounds are inactive against Central Nervous System (CNS), the number of non-conjugated amine groups, computed dipole moments, Protein Interfaces Surfaces and Assemblies (PISA), amidine, guanidine groups, carboxylic acid groups and non- conjugated amide groups were all in recommended ranges except 157232835 with 3 excess non-conjugated amine groups, CHEBI compounds, cidofovir and 49866943 had excess acidic groups while g0mol79715 had exceptionally high number amide groups. The number of reactive functional groups was acceptable except CHEBI compounds had a slightly higher number, these groups may result in false positives in HTS assays and decomposition, reactivity, or toxicity in vivo. The molecular weight of all TMPK Hits was in range while some D13 compounds like Rifampicin had higher values above 725 g. The SASA, FOSA, FISA and Hydrogen acceptors of CHEBI compounds and g0mol79715 were above the recommended values for drug like molecules and these hits may be less stable. Compounds 11986, 11987 and 11988 had higher QPlogPo/w, lower QPlogS, CIQPlogS and QPlog HERG, hence they would have relatively poor absorption/permeation while all other compounds had values in the acceptable range [36]. Some compounds including Cidofovir had poor QPPCaco and QPPMDCK values and this means that the compounds will not be efficiently transported between gut/blood and brain/blood hence lower oral bioavailability, a case already reported for Cidofovir, it’s also toxic to the kidney [8,37]. 11986, 11987, 11988 and g0mol79715 require metabolic steps above 8 for their metabolism. The values for the rule of Five and rule of 3 were all in the recommended range [38]. The difference in ADMET profiles between TMPK and D13 compounds is because D13 compounds are larger due to an extended binding F-Ring in the D13 trimer.
Molecular dynamic simulations
Simulation of 4 apo-proteins showed that our models were all valid and suitable to be used for drug screening purposes while simulation of 20 protein-ligand systems (5 for each protein) shows that the ligands presented are potential inhibitors of respective proteins by forming stable complexes (Figure 4).

Figure 4: The details of molecular interactions between four ligands and MTMPK taken in the middle of the simulation, the Hydrogen bonds are presented as yellow dashed lines, Ligands are colored cyan by element, interacting residues are colored hot pink and the rest of the molecule is a cartoon.
RMSDs of all systems show that all the simulated complexes attained stability over simulation time. The RMSD variation patterns of studied MPXV models are similar to their templates, validating the quality of the models used (Figure S9). The differences in observed RMSDs between the complexes and their corresponding TMPK proteins is a ligand-induced change. For MTMPK, ligands NPC79715 and NPC244454 attained stability at relatively higher RMSDs than other ligands (Figure 5), NPC244454 also had a larger average distance from residues over simulation as well as a smaller number of contacts to the target as compared to other ligands for first 20ns of simulation hence relatively less stable than other ligands in the MTMPK active site (Figures S10 and S11) [39].

Figure 5: The results of molecular simulation dynamics of MTMPK. (A) Shows Protein RMSD, (B) Shows Complex RMSD, (C) Shows Ligand RMSD, (D) Shows RMSF of complexes.
The NPC275538 complex was the most stable over simulation and also supported by maximum Hydrogen Bonds formed and the highest number of contacts with the target as well as the smallest minimum distance from the target over the simulation (Figure S12). Ligand 447688 was less stable compared to other ligands simulated in the VTMPK cavity, it also had the minimum number of contacts with VTMPK over simulation as well as a longer average distance from man-interacting residues over simulation.
All simulated MD13 systems were stable. The ligand RMSD of 11988 from the VD13 complex was higher than that for other ligands over simulation, this is also supported by fewer hydrogen bonds and longer minimum distance from the protein over simulation as well as the lowest number of contacts with the target over simulation.
The major interacting residues have minimum RMSF values over simulation time when compared to the RMSF of ligand- free proteins justifying their stability and interactions with the hits while the RMSF of non-interacting residues shows relatively higher fluctuations. RMSF results also show that the MTMPK loop of residues 26-33 was less stable for complexes of NPC275538, NPC79715 and NPC473924 as compared to other TMPK complexes (Figure 6) [40,41].

Figure 6: The details of the molecular interactions between some of the ligands and VTMPK taken in the middle of the simulation, the Hydrogen bonds are presented as yellow dashed lines.
Average intra-hydrogen Bonds for the model proteins were equal to those of their templates justifying their stability (Figure S14). TMPK H-Bonds ranged 3-9 for all simulated ligands NPC275538 and NPC244454 formed the maximum number of Hydrogen bonds (9) with MTMPK and VTMPK respectively showing significant interactions between them and their targets (Figures 7-10). This also agrees with the fact that these ligands had stable RMSDs over simulation). The D13 H-Bonds ranged from 2-10. The highest number of bonds with D13 was formed by 135566871 (10 for VD13 and 9 for MD13) and the same ligand had stable RMSDs over the simulation as well as shortest minimum distances from the targets compared to other hits and with the maximum number of contacts to the target over simulation (Figures 5,7,9 and 11) (Figure S11).

Figure 7: The details of molecular interactions between some of the ligands and MD13 taken in the middle of the simulation, the Hydrogen bonds are presented as yellow dashed lines, the protein is colored by chain and each color represents a chain.

Figure 8: The results of the VTMPK simulation. (A) Shows protein RMSD, (B) Shows complex RMSD, (C) Shows ligand RMSD, (D) Shows RMSF of complexes.

Figure 9: The simulation results of MD13 systems showing the stability of all systems. (A) Shows protein RMSD, (B) Shows complex RMSD, (C) Shows ligand RMSD, (D) Shows RMSF of complexes.

Figure 10: The details of the molecular interactions between some of the ligands and VD13 taken in the middle of the simulation, the Hydrogen bonds are presented as yellow dashed lines, the protein is colored by chain and each color represents a chain.

Figure 11: The summary the main findings from the molecular dynamic simulation of VD13 systems, (A) Shows protein RMSD, (B) Shows complex RMSD, (C) Shows ligand RMSD, (D) Shows RMSF of complexes.
The Rg of all simulated systems including Apo-Proteins were relatively constant with average values of 3.4 ± 0.015 nm for D13 systems and 1.61 ± 0.025 nm for TMPK systems showing that all simulated systems were compact throughout the simulation and hence stable. The D13 Values are approximately twice those of TMPK due to a larger size of the trimeric D13 compared to the TMPK monomer [41,42]. The compactness hence the stability of all systems gradually increased over simulation evidenced by a very gradual reduction in gyration radius over time. The rate of reduction of gyration radius was higher in the larger trimeric protein than the TMPK hence more conformational changes occurred.
The higher values of D13 SASA compared to TMPK are due to the difference in their protein volumes. The SASA of TMPK reduces at a very small rate over the simulation time compared to that of D13, the reduction of SASA shows an increase in compactness and therefore stability of all the systems. The nearly constant SASA values of TMPK may also be attributed to the presence of positively charged Mg2+. The similar trends of SASA and Rg predict the correctness of molecular dynamics simulation results.
The minimum distances between c-a atoms and ligands over simulation were computed, all minimum distances were between 0.33 and 0.10 nm and therefore all were full filling the distance required for the formation of classic hydrogen Bonds (Figures S12,S13,S14,S15 and S16). The number of contacts between the Ligands and their targets was also computed to further asses the possibility of hydrogen bond formation and stability (Figure S11). When physical properties of potential energy, temperature, density and volume were computed over simulation to monitor the correctness of the process, they were all constant hence validating the correctness of the simulations (Figures S17-S19). The potential energy of all systems was constant over simulation ranging from -1.9E5 KJmol-1 to -1.8E6 KJmol-1, showing stable systems over simulation. The variation of temperature, potential energy, density and volume over simulation (Figures S17,S20).
Essential dynamics, UNI_GBSA and dynamic cross- correlation
Standard ED was conducted on energy and structural data for the last 30 ns of trajectories on only alpha carbons to assess the conformational subspace of the complexes and distinguish the different parts of the energy landscape sampled during the MD simulation to comprehend the dynamic behavior of TMPK and D13 complexes and apo-proteins. The compounds encompassed relatively small subspaces and characterized conformational clusters. Plotting the graph between the two eigenvectors 1 and 2 revealed the type of motion and displacement of atomic fluctuation between the complexes. Subsequent eigenvalues were in decreasing order while the initial few eigenvalues had greater values and their overall direction of motion has been explicated by Principal Components (PCs), as expected, the first PCs explain most of the energy (Figure S21).
All MTMPK complexes showed PC2 ranging from ̴ 2 to ̴ 1 nm² and PC1 between ̴ 1.5 and ̴ 1.5 nm² (Figure 12). NPC79715 showed a shift in PC2 to the negative side. NPC275538 had a unique cluster to the positive side of PC2. The complexes also occupied a small subspace compared to the apo-protein, suggesting more stabilized complexes. The VTMPK PC1 varied from ̴ 1.5 and ̴ 2 nm² while PC2 from ̴ 1 to ̴ 1.5 nm². Ligand 447688 shows a more displaced cluster in the positive direction of PC2 compared to others, hence a better energy distribution.

Figure 12: The results of PCA analysis for all the simulated systems showing the dynamic energy fluctuation plotted between two eigenvectors 1 and 2 generated for the docked complexes showing conformational space of Cα-atoms.
The MD13 PC2 varied from ̴ 4 to ̴ 3 nm² while PC1 from ̴ 5 to ̴ 6 nm², g0mol79715 and CHEBI: 2347 occupied a relatively small range occupancy cluster range compared to other complexes (Figure 12). The Vd13 PC2 ranged from ̴ 4 and ̴ 4 nm² while PC1 from ̴ 5 to ̴ 7 nm², Complexes occupied smaller restricted space compared to the apo-protein suggesting they are more stable and leading to well-defined internal motion behavior vital for complex stabilization (Figure 12). Although most D13 inhibitors had broader energy distribution and some undergo relatively similar conformational stability as apo-proteins, they are still potential morphogenesis-inhibiting compounds.
To further validate the results Molecular simulation dynamics study, we have carried out the MM-GBSA analysis using the uni GBSA tool to estimate the average binding free energy of simulated hit complexes. The results highlight that MM-GBSA analysis (Δ Gbind) correlates well with molecular docking studies. All energies were negative indicating strong binding of proteins and ligands (Tables 2 and 3).
| Ligand | Energy (Kcal/mol) | |
|---|---|---|
| MTMPK | VTMPK | |
| 448657 | -46.994 | - |
| NPC244454 | -48.445 | -49.754 |
| NPC275538 | -52.908 | -51.968 |
| NPC473924 | -55.926 | -47.394 |
| NPC79715 | -48.981 | - |
| 447688 | - | -46.656 |
| 447793 | - | -56.027 |
Note: MTMPK: Monkeypox Thymidylate Kinase; VTMPK: Vaccinia virus Thymidylate Kinase. (-) means that the ligand was not simulated for that target.
Table 2: The results of MM/GBSA for TMPK proteins.
| Ligand | Energy (Kcal/mol) | |
|---|---|---|
| MD13 | VD13 | |
| CHEBI426 | - | -52.745 |
| CHEBI2347 | -70.778 | -68.375 |
| 11987 | -89.567 | - |
| 135566871 | -67.104 | -82.372 |
| 157232835 | -34.319 | - |
| g0mol7915 | -48.982 | -76.654 |
| 11988 | - | -67.789 |
Note: (-) means that the ligand was not simulated for that target.
Table 3: The results of MM/GBSA for D13 proteins.
When the Dynamic Cross-Correlation (DCCs) of apo-proteins are compared to the complexes, it is evident that interacting residues have increased positive correlation, validating their interactions with the suggested targets.
The binding of ligands to MTMPK increased negative cross- correlation peaks for protein regions between the second and ninth α helices and fifth β-sheet compared to a weaker negative correlation between them in the apo-protein, except for ligand 448657 which exhibited a somewhat favorable association in the same areas. Active site residues also show an increased negative correlation between the third β-sheet, fifth and sixth α-helices except for NPC275538 which shows a moderate positive correlation in the same regions (Figure S22). Ligand NPC244454 increases positive cross-correlation peaks between different regions of VTMPK followed by 447793 while the other 3 ligands show an overall increase in weak to moderate negative cross-correlations between different domains of the protein as well as between active site residues and the highly flexible C-terminus loop (Figures S23 and S24).
In this study, we modeled MPXV protein structures using VACV templates, they were refined and their qualities were validated. They were then screened against 602,413 small molecules using pharmacophore and molecular docking methods and their binding affinities were determined. Qikprop ADMET properties were also computed and all TMPK compounds had good ADMET profiles compared to those of D13 proteins which may require a more intense lead optimization for their further applications. These compounds had very good docking scores and Binding affinities to our targets compared to the used standards of Cidofovir and Rifampicin. Finally, their best-ranking complexes were taken for MD studies to further assess their stability.
The MD RMSDs showed that all studied complexes were stable over simulation. The ligands also formed reasonable H-Bonds with their respective targets over simulation. The same outcome was predicted by the SASA and Radius of Gyration graphs and the RMSF projected that the ligand's binding to the protein molecules was effective. The results of the docking and simulation procedures were supported by the computed MM/GBSA energy, which was negative and indicated strong binding. Further PCA analysis and DCC calculation indicated that all formed complexes were stable with ligands NPC275538, NPC244454, 135566871 and CHEBI compounds being superior to other Hits. The findings indicate that all of the molecules investigated here compete with one another for the binding sites for TDP and Rifampicin in the TMPK and D13 proteins, respectively. Since the studied proteins are highly conserved among OPVs, these compounds can be potential inhibitors for all this group of viruses hence attracting polypharmacology studies. As a result, these molecules should be put through experimental testing to confirm their antipox potential. This finding, therefore, makes a substantial contribution to the development of novel medications for the treatment of monkeypox since there are no authorized MPXV antivirals.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
Citation: Charles S, Edgar MP, Kasoma NA (2023) The Hunt for Antipox Compounds against Monkeypox Virus Thymidylate Kinase and Scaffolding Protein Leveraging Pharmacophore Modeling, Molecular Docking, ADMET Studies and Molecular Dynamics Simulation Studies. Virol Myco. 12.280.
Received: 03-Oct-2023, Manuscript No. VMID-23-27288; Editor assigned: 05-Oct-2023, Pre QC No. VMID-23-27288 (PQ); Reviewed: 19-Oct-2023, QC No. VMID-23-27288; Revised: 26-Oct-2023, Manuscript No. VMID-23-27288 (R); Published: 02-Nov-2023 , DOI: 10.35248/2161-0517.23.12.280
Copyright: © 2023 Charles S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.