Journal of Clinical Trials
Open Access
ISSN: 2167-0870
ISSN: 2167-0870
Review Article - (2012) Volume 2, Issue 2
Post operative nausea and vomiting (PONV) are common complications and occurs in as many as 70%- 80% of high risk surgical patients. The latest prophylactic treatment recommended in the Society of Ambulatory Anesthesia Guidelines (SAMBA) for the management of Post operative Nausea and Vomiting for high risk patients is a combination of 2 or more interventions (multimodal therapy). A combination of a 5-HT3 receptor antagonist with Dexamethasone and/or Droperidol, or a 5-HT3 receptor antagonist with Droperidol alone, or Dexamethasone with Droperidol, have been the pharmacologic combination therapies suggested in these guidelines. Scopolamine “Transdermal Scop” is a belladonna alkaloid with anticholinergic properties. It acts as a nonselective muscarinic antagonist, approved by the FDA for PONV prophylaxis. The use of this novel drug in a triple therapy combination with Dexamethasone and/or Droperidol could be an effective treatment for the prevention of PONV. However, since the FDA issued a warning stating that Droperidol may cause life – threatening arrhythmias as well as a prolongation of the QTc interval, the need to discover new combination therapies for PONV prevention in high risk patients is still in demand. Therefore, we hypothesize that the use of a triple prophylactic therapy consisting of Scopolamine, Dexamethasone, and Ondansetron will be an effective treatment for the prevention of PONV in patients at a high risk for developing PONV during the first 120 hours after neurosurgery.
Postoperative nausea and vomiting (PONV) is one of the most common complaints concerning both patients and clinicians [1]. Prevention and treatment of PONV are key patient care issues that greatly affect comfort and satisfaction during the postoperative period. In a survey of 101 patients, the most undesirable surgical outcome reported was vomiting, ranking even higher than pain [2]. PONV alone is never fatal, but it can result in significant complications including: aspiration, pneumonitis, dehydration, wound dehiscence, acid-based disorder and electrolyte imbalance, hematoma formation, esophageal rupture, and increases in intraocular and intracranial pressures due to acute blood pressure elevations [3-5].
According to the Society for Ambulatory Anesthesia (SAMBA) Consensus Guidelines for the Management of PONV, the incidence of PONV is between 20-30% of all patients undergoing surgery [6]. In absence of prophylactic treatment, PONV occurs in the 70%-80% of high risk patients undergoing surgery [6]. Incidence of PONV in neurosurgery is analyzed separately, with 50% of patients experiencing nausea and 39% of patients vomiting [7]. In a study of postoperative complications, nausea and/or vomiting was the most common complication observed in the first 4 hours in 39 % of patients following neurosurgery [8]. During the first 48 hours following neurosurgery, an even higher prevalence of nausea and vomiting, 70% and 55% respectively, was demonstrated in patients given a placebo with no prophylactic anti-emetic treatment prior to undergoing supratentorial craniotomies [4].
Three categories of baseline risk factors, patient specific, anesthetic, and surgical, are independent predictors of PONV to aid clinicians in determining which patients are candidates for prophylaxis. The most prevalent patient-specific risk factors for PONV are females, nonsmokers and history of PONV or motion sickness [6]. Other potential risk factors include history of migraines, young age, anxiety, and a low American Society of Anesthesiologists (ASA) physical status classification [6]. Anesthetic risk factors include the use of volatile anesthetics during general anesthesia, use of nitrous oxide, and use of postoperative opioids [6]. Surgical risk factors are related to the duration and type of surgery. Each 30-minute increase in duration increases PONV risk by 60%, so that a baseline risk of 10% is increased to 16% after 30 minutes. Type of surgery with a high incidence of PONV include: laparoscopy, laparotomy, breast, strabismus, plastic, maxillofacial, gynecologic, abdominal, ophthalmologic, urologic and neurologic surgeries [6].
A single risk factor for PONV is not sensitive or specific enough to be used to assess the risk of PONV. According to the simplified risk score model from Apfel et al [9], the greater the number of independent predictors, the higher the risk for PONV. Each factor has a punctuation value of 1, which added will equal 0 – 4. When 0, 1, 2, 3, or 4 of the depicted independent predictors are present; the corresponding risk for PONV is approximately 10%, 20%, 40%, 60%, or 80% [6].
The presence of 1 risk factor correlates with 20% risk for PONV, as each subsequent risk factor is added, risk increases by 20%, resulting in an 80% risk if all 4 risk factors are present [9]. The SAMBA Consensus Guidelines for the Management of PONV include assessing the patient’s risk for PONV, reducing the baseline risk factors for PONV, and providing prophylactic treatment. Some strategies to reduce the baseline risk for PONV include avoidance of general anesthesia, use of Propofol for induction and maintenance of anesthesia, avoidance of Nitrous Oxide and other volatile anesthetics, minimizing the use of opioids during and after the surgery, minimizing the use of Neostigmine and adequate hydration [6]. However, these PONV risk elevating techniques cannot be avoided during this type of neurological surgery.
While prophylactic treatment is not recommended for all patients, it should be used when a patient’s risk of developing PONV is adequately high. Situations when it is advantageous to prevent vomiting can be when patients have wired jaws or increased intracranial pressure [6]. SAMBA Consensus Guidelines for the Management of PONV suggest the use of several drugs combination for the prophylaxis of PONV [6].
- 5-hydroxytryptamine (5-HT3) recepytor antagonists: Ondansetron, Dolasetron, Granisetron, Tropisetron and Palonosetron
- Corticosteroids: Dexamethasone
- Dopamine (D2) receptor antagonists: Droperidol and Haloperidol
- Antihistamine drugs: Dimenhydrinate
- Anticholinergic drugs Scopolamine
Unfortunately, no single drug is completely efficacious in patients at high risk for PONV [5]. The SAMBA guidelines recommend the use of one or two antiemetic combinations in adults at moderate risk; and two or more combinations for adults at high risk [6]. It is specifically, recommended to administer 2 or more prophylactic antiemetic drugs from different classes to patients who have high risk for PONV [6]. Vomiting can be induced through multiple pathways which ultimately activate the vomiting center in the lateral reticular formation of the medulla. Activation of this center leads to the visceral and motor output involved in vomiting [10]. Since there are multiple pathways of activation, a multi-modal approach should be used to prevent PONV [11]. Correspondingly, it has been proven that a combination multi-modal prophylactic therapy is more effective at preventing PONV than single drug therapy [11]. Furthermore, in using a combination of drugs, the dosage of each drug could be reduced, in the effort to decrease side effects [11]. Overall, the guidelines state that patients at moderate risk should receive one or more prophylactic drugs from different classes and those at high risk, two or more [6].
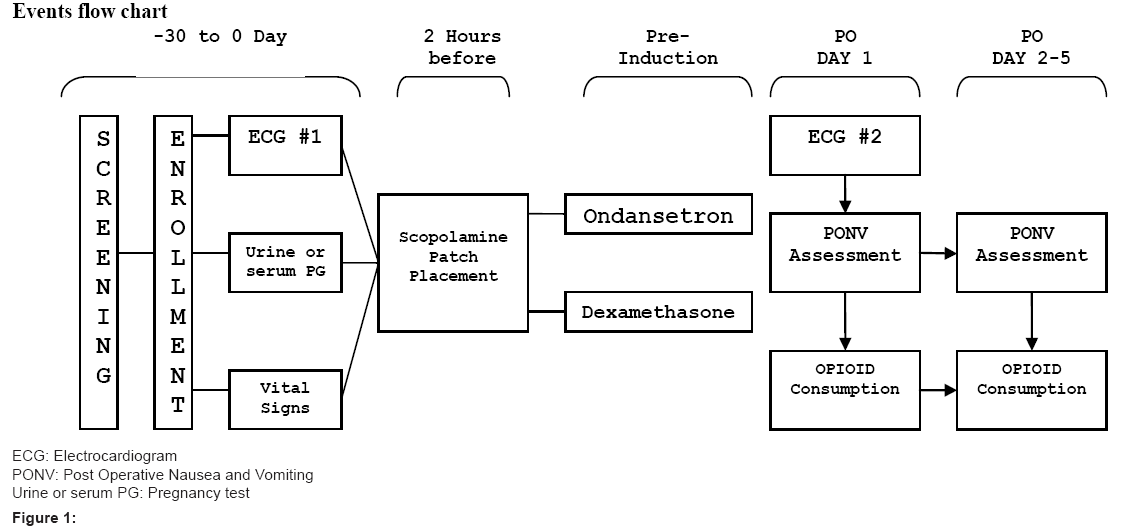
Figure 1: Events flow chart.
Schedule of observations and procedures
| Screening (-30 Days) | Baseline Time 0 | 2 Hours Before Surgery | Pre- Induction | PO Day 1 | PO Day 2 - 5 | |
|---|---|---|---|---|---|---|
| Informed Consent & HIPAA | X | X* | ||||
| Inclusion & Exclusion Criteria | X | X* | ||||
| Demographics | X | X* | ||||
| Medical History | X | X* | ||||
| Vital Signs | X | |||||
| ECG | X | X | ||||
| Urine or serum Pregnancy Test | X | |||||
| Opioid Consumption | X | X | X | |||
| Scopolamine Patch Placement | X | |||||
| Dexamethasone and Ondansetron administration | X | |||||
| Adverse Events | X | X | X | X | ||
| Concomitant Medication | X | X | X | X |
Table 1
The guidelines emphasize the use of the following combination therapies in adults: Droperidol and Dexamethasone, 5-HT3 receptor antagonist and Dexamethasone, 5-HT3 receptor antagonist and Droperidol, or a triple-therapy with a 5-HT3 receptor antagonist, Dexamethasone, and Droperidol [6,12]. Droperidol is no longer recommended as prophylactic treatment for PONV because of a recent FDA black box warning highlighting possible prolongation of the QT interval and dangerous heart arrhythmias.
In effort to find an alternative to Droperidol in triple-therapy treatment for PONV, Scopolamine may provide effective nausea and vomiting prophylaxis. Recently approved by the FDA, the Transdermal Scopolamine patch has shown efficacy in the prevention of nausea and vomiting associated with recovery from anesthesia and surgery used as a single drug. However the combined use of Scopolamine, Dexamethasone and Ondansetron may be a more effective method of treating patients with a high risk of PONV. Scopolamine inhibits muscarinic receptors for acetylcholine and acts as nonselective muscarinic antagonists, producing both peripheral antimuscarinic properties and central sedative effects, as antiemetic and amnestic effects as well [12,13]. The Transdermal Scop system consists of a circular flat patch designed for continuous release of scopolamine following application to the mastoid area. Each system contains 1.5 mg of scopolamine base, and it is programmed to deliver 1.0 mg of scopolamine at an approximately constant rate to the systemic circulation over 3 days. After removal of the used system, there is some degree of continued absorption. Scopolamine is well-absorbed percutaneously, following application circulation plasma levels are detected within 4 hours with peak levels within 24 hours [13,14].
Transdermal Scopolamine patch was found to be significantly superior to Ondansetron or Placebo as a prophylactic therapy for prevention PONV in a study performed by Harnett et al. in 240 females undergoing cesarean delivery under spinal anesthesia and opioids analgesia. The results showed that the overall rates for all emesis were reduced to 40 % in the Scopolamine group [15]. Another trial examining Scopolamine versus Placebo in 48 patients undergoing gynecologic laparoscopy showed that it significantly reduced in the incidence and severity of nausea and vomiting in the first 24 hours after surgery [16].
The use of Transdermal Scopolamine specifically for the prevention of PONV has also been recently examined. A study by Sah et al. [17] examined active Transdermal Scopolamine patch plus Ondansetron compared to Placebo patch plus Ondansetron as a prophylaxis treatment for PONV in 126 patients undergoing plastic surgery. This study found a statistically significant reduction in PONV between 8 and 24 hours after surgery in patients receiving Transdermal Scopolamine plus Ondasetron. An additional study performed by Shari et al. [18] examined active Transdermal Scopolamine patch plus Ondansetron compared with Placebo patch plus Ondansetron as a prophylaxis treatment for PONV in 56 high risk patient’s scheduled to receive general anesthesia for no longer than 1 hour. This study also found significant reduction in PONV using a combination of Transdermal Scopolamine and Ondansetron.
In 2009, a study by Gan et al. [19] confirmed the previous findings. The study examined active Transdermal Scopolamine patch plus Ondansetron compared with Placebo patch plus Ondansetron as a prophylaxis treatment for PONV in 620 females patients undergoing laparoscopic or breast augmentation surgery. This study found significant reduction in PONV 24 hours after surgery in the group who received Transdermal Scopolamine plus Ondansetron when compared to the group received Placebo patch plus Ondansetron. All of these studies found significant reduction in adverse events in the group receiving Transdermal Scopolamine plus Ondansetron. Altogether, these studies indicate that Transdermal Scopolamine in addition to Ondansetron may benefit patients at high risk for PONV.
In OSU all patients undergoing Neurological Surgery received Ondansetron considered as standard of care. In this study, triple therapy consists in Scopolamine, Ondansetron and Dexamethasone. Each drug has been individually proved to help preventing PONV. Based on previous studies results showed that single therapy for PONV prophylaxis has an average of 30 % of complete antiemetic response and combination therapy have an average of 40-60 % of complete antiemetic response. Therefore we hypothesized that the effects of the combination therapy are going to be more satisfactory compared with single therapy.
Is triple-therapy with Scopolamine, Ondansetron and Dexamethasone effective in preventing post-operative nausea and vomiting in high risk patients undergoing neurological surgery under general anesthesia?
The use of a prophylactic triple-therapy consisting of Scopolamine, Ondansetron and Dexamethasone, will be an effective treatment for the prevention of PONV in high risk patients undergoing neurological surgery under general anesthesia during the first 120 hours postoperative at The Ohio State University Medical Center (OSUMC).
Primary objective
- To assess the efficacy of triple therapy with Scopolamine, Ondansetron and Dexamethasone for prevention of post operative nausea and vomiting in high risk patients during the first 24 hours after neurological surgery under general anesthesia
Secondary objective
- To assess the efficacy of triple therapy with Scopolamine, Ondansetron and Dexamethasone for prevention of post operative nausea and vomiting in high risk patients during a delayed period after neurological surgery under general anesthesia
Primary endpoint
- Proportion of patients with a complete response/complete control during the first 24 hours after neurological surgery under general anesthesia
- Assess the severity of nausea and vomiting during the first 24 hours after neurological surgery
Secondary endpoint
- Proportion of patients with a complete response/complete control during a delayed period (between 24 and 120 hours) after neurological surgery under general anesthesia
- Assess the severity of nausea and vomiting during a delayed period (between 24 - 120 hours) after neurological surgery under general anesthesia
- Assess the time to treatment failure
- Assess the time to first emetic episode
- Assess the time to significant nausea
The following definitions will be used:
- Complete Response: No emetic episode and no rescue medication
- Complete Control: No emetic episode, no rescue medication, and no more than mild nausea
- Mild nausea: Nausea rated ≤ 4 on a 0 to 10 verbal response scale or nausea that required rescue therapy
- Treatment failure: Time to first emetic episode and/or to first use of rescue medication
- Significant Nausea: Nausea rated ≥ 4 on a 0 to 10 verbal response scale or nausea that required rescue therapy
Study population
Adult patients, at OSUMC, 18 to 85 years of age, with an American Society of Anesthesiologists (ASA) physical status of I to III who are scheduled to undergo neurological surgery under general anesthesia requiring opening of the cranium and dura mater.
Sample size
40 subjects who give written informed consent to participate in the study and who meet all inclusion and no exclusion criteria will be included in a single treatment group.
Statistical method
The efficacy data will be analyzed based on the intention-to-treat principal. The intent-to-treat cohort will consist of all patients given triple therapy with Scopolamine, Ondansetron and Dexamethasone with at least one post operative efficacy assessment. The safety cohort will consist of all patients given triple therapy with Scopolamine, Ondansetron and Dexamethasone who had at least one post dose assessment. Patients who withdraw from the study will be considered non-responders on and after the day of withdrawal. Baseline patient demographics, clinical characteristics, and safety data will be summarized with descriptive statistics, such as mean, median, standard deviation, etc. The proportion of patients experiencing a complete response (no emesis and no rescue medication required) will be evaluated at 24 - hour intervals from 0 - 120 hours post operatively, including the acute (0 - 24 hours), delayed ( > 24 - 120 hours), and overall (0 - 120 hours) intervals. Efficacy data will be summarized using descriptive methods with confidence intervals determined for mean values and proportions. We will examine the comparability of the treatment group with respect to important preoperative factors. Logistic regression will be used to test the primary hypothesis with demographic characteristics as potential covariates in the model. For the number of rescue therapy treatments used during the 24, 48, 96, 72 and 120 hour postoperative period, Chi-square or exact test for proportions will be performed. Adjustments for multiple comparisons will be performed (Bonferroni or Holm’s methods). For other secondary endpoints, the analysis plan will be similar to the one for the primary endpoints.
The desire sample size for the study is 40 patients and it is based on the hypothesis of the primary aim. This sample size is determined to achieve 80 % power to detect an increase (P1-P0) of 20 % in the proportion of patients with complete response using a one-sided binomial test at the significance level of 0.05. This sample size will allow us to construct 95% confidence interval with limits of 0.47 to 0.77. We will seek n=40 subjects to account for screening and attrition in the study. Based on the literature (Gan et al, 2009) the proportion of patients with complete response was assumed to be 60 % under the null hypothesis. This sample size assumes a 10 % dropout rate.
Inclusion criteria: Subjects must meet ALL of the criteria below for entry.
1. Male or female, 18 to 85 years of age
2. Subject’s American Society of Anesthesiologist (ASA) physical status of I to III who are scheduled to undergo neurological surgery requiring opening of the cranium and dura matter under general anesthesia
3. Subjects able to provide written informed consent to participate in the study
4. An expected post operative hospitalization stay of at least 24 hours
5. Subject’s surgery is expected to require at least 1 hour of general anesthesia
6. Female subjects who have a negative urine or serum pregnancy test within 1 day of surgery or who have been surgically sterilized or are postmenopausal
Exclusion criteria: Subjects will NOT be eligible for entry into the study if any of the following criteria are identified.
1. Subjects who are prisoners
2. Subjects who has limited decision making capacity or lack the ability to consent
3. Subjects with a history of alcohol abuse or drug abuse within last year
4. Subjects with a history of allergies reaction to, intolerances of, or contraindications for any of the study medications
5. Females who are pregnant or are breastfeeding
6. Subjects who have had retching/vomiting or moderate to severe nausea in the 24 hours prior to anesthesia or suffer chronic nausea and/or vomiting
7. Subjects who have been treated with any drug or other treatment with antiemetic efficacy within the last 24 hours prior to the start of treatment
8. Subjects who have participated or are currently participating in a clinical trial of an investigational drug within 30 days prior to surgery
9. Subjects with a history of acute narrow-angle glaucoma (treated or untreated increased intraocular pressure), gastrointestinal obstruction, or urinary tract obstruction
10. Subject with a history of latex allergy
11. Subjects that have received Transdermal Scop within the past 30 days
12. Any condition, which in the opinion of the investigator would make subject ineligible for participation in the study such as history of unstable cardiovascular, pulmonary, renal, hepatic, neurologic (seizures), hematologic or endocrine abnormality
13. Subjects with a history of malignant hyperthermia
14. A cancer patient who has had chemotherapy within 4 weeks prior to the screening visit
15. Any kind of emetogenic radiotherapy within 8 weeks prior to the screening visit
This is a single-center, prospective, non-randomized, openlabel, single-arm study involving 40 subjects at the OSUMC who are scheduled to undergo neurological surgery under general anesthesia requiring opening of the cranium and dura mater. Eligible subjects that provide voluntary and written informed consent will be included in one single treatment group.
Subjects will undergo screening procedures within 30 days prior to administration of study treatment. The research staff will collect the subject’s medical history (including reason for surgery, scheduled surgical procedure, anesthesia modality and surgery scheduled length), demographics (including gender, age, race, and ethnicity), history of alcohol or drug abuse and history of PONV/motion sickness, smoking, and allergies.
Prior to surgery, vitals and study procedures, including ECG and urine or serum pregnancy test will be performed. Patients will receive an active Transdermal Scop 1.5 mg scopolamine patch over the mastoid area within 2 hours prior to surgery. All subjects will receive Ondansetron 4 mg Intravenous (IV) as a single dose and Dexamethasone 10 mg IV as a single dose, both to be administered at induction of general anesthesia.
The transdermal scopolamine patch will remain in place until 24 hours after surgery. If the patch becomes dislodged from the skin at any point during the 24 hour period, the attending Anesthesiologist will reapply it on the same site and cover it with a protective bandage.
Anesthesia and procedure start and end time, will be recorded. Admission and discharge time of the subject to the post-anesthesia care unit (PACU), surgical intensive care unit (SICU) and nursing floor will be recorded.
Nausea and vomiting will be assessed every 24 hours for 5 days via direct subject interview and chart review. If the subject is discharged before the end of the 5 day time period, the subject will then be contacted via phone call by study personnel to complete assessments. Episodes of nausea, vomiting and administration of rescue therapy for either nausea or vomiting will be recorded. In addition, the severity of each nauseous or emetic episode will be recorded. Nausea will be rated by the patient using a verbal response scale (0-10). Vomiting will be evaluated by the investigator or nursing staff numerically as either 0 (no vomiting), 1 (mild vomiting:vomiting is anticipated or 1-2 episode in 12 hours,small amount of emesis), 2 (moderate vomiting:3-5 episodes in 12 hours, breakthrough vomiting) or 3 (severe vomiting:6-7 episodes in 12 hours, intractable, incessant, projectile). Following the first 24 hours after administration of the prophylactic triple therapy an ECG will be performed.
Medication with antiemetic properties, intraoperative medication and opioids daily consumption will be recorded during the hospital course.
Rescue medication
Ondansetron 4 mg IV will be administered as rescue medication in the event of intractable nausea, emetic episode, or at subject request if agreed upon by the treating medical staff, surgeon or principal investigator, in accordance with the standard operating procedures at OSUMC. In the event that the second dose of intravenous Ondansetron was unsuccessful in controlling the PONV, any antiemetic medication may be administered per standard of care.
Patients with intractable post operative nausea and vomiting will have a nasogastric tube inserted into their stomach in accordance with standard of care practices at OSUMC.
Efficacy assessments
Efficacy parameters will be collected at each 24 hour time interval throughout the 120 hours of the post operative period include the number and severity of emetic episodes experienced, the use of rescue medication, and the intensity of nausea experienced post operatively. Nausea severity will be assessed with the use of a verbal response scale on a 0 to 10 verbally elicited scale. The 11 point categorical scale to be used to rate severity of nausea ranges from 0 (no nausea) to 10 (nausea as bad as it could be). Vomiting will be evaluated by the investigator or nursing staff numerically as either 0 (no vomiting), 1 (mild vomiting), 2 (moderate vomiting) or 3 (severe vomiting).
Primary efficacy assessment
The percentage of patients with no emetic episodes over 0 – 24 hrs post operatively
Secondary efficacy assessment
-No emetic episodes for the following time intervals: 24-48, 24-72, 24-96 and 24-120 hours
-Number of rescue therapy treatments administered over 0-24, 0-48, 0-76, 0-96 and 0-120 hours
-Percentage of patients achieving complete response over 0-48, 0-72, 0-96 and 0-120 hours
- Percentage of patients with no nausea over 0-48, 0-72, 0-96 and 0-120 hours post operatively
- Time of first rescue medication
- Time of first emetic episode
-Time to significant nausea
-Number of emetic episodes at 0-24, 0-48, 0-72, 0-96 and 0-120 hours
Safety assessments
Safety parameters will be assessed by monitoring vital signs, adverse events, assessing baseline and 24 hour ECG and urine or serum pregnancy test. A physical exam will be performed at screening.
The occurrence of adverse events (AE) and serious adverse events (SAE) will be recorded during the 5 day post operative period. For each adverse event the relationship to the study medication, severity, expectedness of an adverse event and outcome will be determined by the Principal Investigator and recorded in the study source accordingly.
All study medication and study procedures required by protocol that are not considered as part of the standard of care, and that will be obtained solely for research purposes, will be provided by the study at no cost to the subjects. In the case a subject withdraws from the study because of a serious adverse (SAE) local IRB will be notified within 10 days.
Adverse event definition
An AE is defined as any untoward medical occurrence in a patient or clinical investigation subject administered a medicinal product and which does not necessarily have to have a causal relationship with this treatment. An AE can be any unfavorable and unintended sign, symptom, abnormal laboratory finding or a temporally disease associated with the use of a study drug, whether or not considered related to the study drug.
Planned hospital admissions and/or surgical operations for an illness or disease that existed before the subject was enrolled in a clinical study are not to be considered AEs.
Serious adverse event
A SAE is any untoward medical occurrence that at any dose:
- Results in death
- Is life-threatening
- Results in persistent or significant disability/incapacity
- Requires in-subject hospitalization or prolongs hospitalization
- Is a congenital anomaly/birth defect
- Is another medically-significant event that, based upon appropriate medical judgment, may jeopardize the subject and may require medical or surgical intervention to prevent one of the outcomes listed above
Withdrawal criteria from the study
According with the declaration of Helsinki, participants have the right to withdraw from the study at any time for any reason. The principal investigator also has the right to remove a subject from the study. Reasons for which a subject may be removed from the study include:
- An adverse event
- The request of the subject, his/her legal representative or caregiver, investigator, whether for administrative or other reasons
- Non - compliance with medication, protocol violation or unreliable, behavior
- Any clinically significant abnormal laboratory values, or other clinically significant abnormalities identified by the principal investigator according to his clinical judgment, will be followed by appropriate tests and/or procedures until these values have returned to normal or to clinically acceptable levels or can be attributed to other causes other than study drug.
The principal Investigator may withdraw an enrolled and treated subject from the study for any of the following reasons:
-Occurrence of a serious or intolerable adverse event
Emergence of a clinically significant change in a laboratory parameter(s)
-T he subject requests to be discontinued from the study
- A protocol violation sufficiently serious as to require subject withdrawal
- General or specific changes in the subject’s condition that render further treatment unreasonable or unsafe within the standards of clinical practice in the judgment of the Principal Investigator or treating physician
Any subject may leave the study at any time. If a subject decides to stop participating in the study, there will be no penalty. The subjects will not lose any benefits to which they are otherwise entitled. Their decision will not affect their future relationship with The Ohio State University