Journal of Clinical Trials
Open Access
ISSN: 2167-0870
ISSN: 2167-0870
Protocol Article - (2015) Volume 5, Issue 3
Background: Hepatitis B surface antigen (HBsAg) clearance marks the prime event of HBV elimination in chronic hepatitis B and is associated with better overall outcome. Despite reliable viral suppression under standard treatment with nucleos(t)ide analogues (NUCs) the goal of HBsAg clearance is rarely achieved. Also synchronous combination of NUCs with pegylated interferon-α-2a (peg-IFNα) has not been superior compared to peg-IFNα monotherapy in prospective randomized trials. However, sequential addition of peg-IFNα to an ongoing NUC regimen has provided higher HBsAg clearance rates in uncontrolled pilot studies.
Methods/Design: In this protocol we investigate the sequential addition of open-label peg-IFNα for 48 weeks to an ongoing NUC regimen following patients written informed consent. Included are patients with HBeAg negative chronic hepatitis B and a suppressed HBV DNA (<20 IU/mL) for a minimum of 12 months under NUCs prior to trial enrollment. Patients are randomized (2:1 ratio) to peg-IFNα add-on/ NUC treatment or a control group receiving continuous NUCs only. Patients are followed regularly during the trial intervention including a per protocol follow-up for 24 weeks after end of treatment. The primary endpoint is the objective response after 48 weeks of combination therapy, defined by a confirmed reduction of HBsAg by ≥ 1log10 IU/mL compared to baseline. Secondary endpoints are the HBsAg seroconversion rate, safety and tolerability of the IFN add-on therapy regimen. The trial was approved by the local ethic committees of all participating study sites.
Trial registration: The trial was registered on the 10th of June 2011 at EudraCT (ID number 2011-002812-10).
Keywords: Hepatitis B; Interferon IFN; HBV surface antigen; HBsAg therapy
AE: Adverse Event; ALT: Alanin-aminotransferase; AFP: Alpha-Fetoprotein; LKM: Anti-Liver-Kidney Microsomal Antibody; AMA: Anti-Mitochondrial Antibody; ANCA: Anti-Neutrophil Cytoplasmatic Antibody; ANA: Anti-Nuclear Antibody; SMA: Anti-Smooth Muscle Actin Antibody; SLA: Anti-Soluble Liver Antigen Antibody; AST: Aspartat-Aminotransferase; BUN: Blood Urea Nitrogen; cHB: Chronic Hepatitis B; eCRF: Electronic Case Report Form; EOF: End of Follow-up; EOT: End of Treatment; fT4: Free Tetraiodothyronine; fT3: Free Triiodothyronine; Gamma-GT: Gamma-glutamyltransferase; GCP: Good Clinical Practice; HCC: Hepatocellular Carcinoma; HBeAg: Hepatitis B e Antigen; HBsAg: Hepatitis B Surface Antigen; HDL: High Density Lipoprotein; HLA: Human Leukocyte Antigen; IgG: Immune Globuline G; INR: International Normalized Ratio; LDH: Lactate Dehydrogenase; LDL: Low Density Lipoprotein; mITT: Modified Intention-to-Treat; NUCs: Nucleos(t)ide Analogues; PCR: Polymerase Chain Reaction; peg-IFNα: Pegylated Interferon-α-2a; PT: Prothrombine Time; SAEs: Serious AEs; SAP: Statistical Analysis Plan; TSH: Thyreoid-stimulating Hormone; ULN: Upper Limit of Normal.
Chronic hepatitis B (cHB) is one of the world leading causes of chronic liver disease, though treatment with nucleos(t)ide analogues (NUC) achieves a reliable suppression of HBV replication and reduces HBV-associated morbidity and mortality. Until now a defined treatment endpoint has not been defined for NUCs, particularly as cHB clearance indicated by hepatitis B surface antigen (HBsAg) loss and anti-HBs seroconversion is rarely achieved. The resulting long-term NUC treatment requires a high compliance by the cHB patient is cost intensive and could provoke unknown long-term toxicities.
Efforts to improve HBsAg clearance by synchronous combination of NUC and pegylated interferon alfa-2a (peg-IFNα) for 48 weeks were not superior compared to peg-IFNα monotherapy. Low HBsAg clearance rates of 3.95% following IFN monotherapy and 2.79% following synchronous IFN/ lamivudine combination were achieved in HBeAg negative cHB patients for example [1-3]. Recent data suggest that a long lasting NUC therapy is able to restore HBV-directed T cell responses, which are essential to prime HBsAg clearance from the circulation [4]. Immune reconstitution indicated by a decrease of dysfunctional T cells hereby requires a long standing NUC treatment and HBV suppression [5].
Following these observations we hypothesized that a stable NUC therapy for more than one year could restore responsiveness to peg-IFNα added to an ongoing NUC regimen. A pilot study investigating the HBsAg decline under IFN add-on therapy in 12 patients was scheduled at our center and showed a complete HBsAg loss in two patients (16.6%) receiving peg-IFNα for a total of 48 weeks. Anti-HBs seroconversion occurred after 32 and 40 weeks in this cohort [6]. An independent non-controlled trial identified favorable HBsAg loss rates of 60% in hepatitis B e antigen (HBeAg) negative patients receiving response-guided peg-IFNα for a maximum of 96 weeks [7]. Following these preliminary data we were encouraged to confirm our findings in a prospective and randomized clinical trial investigating reduction of HBsAg serum concentrations under peg-IFNα add-on therapy for 48 weeks compared to a control group receiving an ongoing NUC monotherapy.
Study design
Peg-interferon ADDed to an Ongoing Nucleos(t)ide treatment in patients with chronic hepatitis B to induce decrease of HBs antigen (PADD-ON) is a prospective randomized open-label multicenter phase IIb clinical trial (EudraCT No: 2011-002812-10).
Objectives and outcomes
The PADD-ON trial investigates the effect of pegylated interferon alfa-2a (peg-IFNα) added to an ongoing antiviral NUC therapy in HBeAg negative cHB patients. The primary endpoint is the objective response after 48 weeks of combination therapy, defined by a confirmed reduction of ≥ 1log10 in HBsAg serum concentrations (IU/mL) compared to baseline. Secondary endpoints include alternative response parameters as well as safety and tolerability of the IFN add-on therapy regimen (Table 1). Patient recruitment has been started in September 2012 and is proposed to end in 2015. Recruitment and treatment of patients is performed in 22 active trial centers in Germany.
| Primary endpoint |
|---|
| Reduction of ≥1log10 IU/mL in HBsAg after 48 weeks of combination therapy compared to baseline. |
| Secondary endpoints |
| Decline of quantitative HBsAg at week 12 and 24 compared to baseline |
| Proportion of patients showing a relative HBs antigen concentration decline by ≥10% between week 0 (baseline) and week 24 |
| Anti-HBs seroconversion defined as percentage of patients who become HBsAg negative and anti-HBs positive during the observation period |
| Safety and tolerability of peg-IFNα when added to a continuing treatment with tenofovir, entecavir, lamivudine, adefovir, or a combination of those: Analysis of adverse events and laboratory data. |
Table 1: Study objectives and endpoints.
The trial adheres to an individual screening period of 4 weeks. After randomisation, the treatment period takes 48 weeks. Follow-up continues for another 24 weeks (Figure 1).
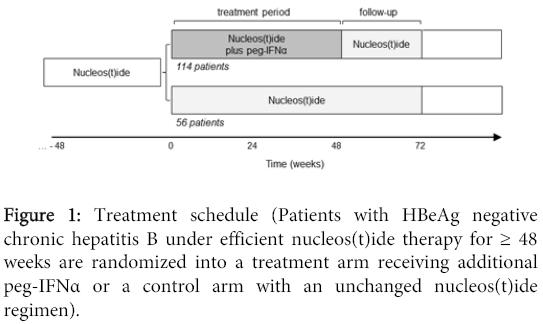
Figure 1: Treatment schedule (Patients with HBeAg negative chronic hepatitis B under efficient nucleos(t)ide therapy for ≥ 48 weeks are randomized into a treatment arm receiving additional peg-IFNα or a control arm with an unchanged nucleos(t)ide regimen).
Selection of study population: HBeAg negative hepatitis B patients, who have shown a continuous serum HBV DNA concentration<20 IU/mL for more than one year under ongoing NUC treatment, are eligible for the PADD-ON trial. Patients are further selected by specific inclusion and exclusion criteria (Table 2). After patients have been informed about the trial, written informed consent in accordance with good clinical practice (GCP) regulations and the local legislation must be obtained prior to any study related procedures. No patient is allowed to enrol in this trial more than once. Included patients who discontinue from the trial before intake of at least one dose of study medication will be replaced.
| Inclusion criteria |
|---|
| Chronic hepatitis B, HBeAg negative, confirmed by documented positive HBsAg for > 6 months |
| Treatment with a nucleos(t)ide regimen (lamivudine, adefovir, entecavir, tenofovir or one of the following combinations: lamivudine/adefovir, lamivudine/tenofovir, entecavir/adefovir or entecavir/tenofovir) and a fully suppressed viral load for at least 12 months (below limit of detection in conventional HBV-PCR assays, e.g. <116 copies/ml or <20 IU/mL) |
| Quantitative HBsAg value available at least 180 days prior to screening |
| HBsAg ≥100 IU/mL |
| Normal retinal finding on fundoscopy within 6 months prior to day 1 (baseline) |
| 18-70 years |
| Ability of patient to understand character and individual consequences of clinical trial |
| Signed and dated informed consent of the patient must be available before start of any specific trial procedures. |
| Women of childbearing potential have to be practicing a medically accepted contraception during trial and a negative pregnancy test (serum or urine) should be existent before trial. |
| Exclusion criteria |
| Patients presenting with any of the following criteria will not be included in the trial: |
| HBeAg positive Hepatitis B |
| Co-infection with HCV, HDV or HIV – as based on positive serology or PCR |
| Ongoing antiviral treatment with Telbivudine |
| Contraindications against treatment with peg-IFNα, e.g. severe depression, epilepsy, autoimmune diseases, pregnancy, leukocytopenia or thrombocytopenia at screening, etc. |
| Preexisting polyneuropathy |
| Decompensated liver disease, or history of decompensated liver disease, as evidenced by ascites, portal hypertension, jaundice or hepatic encephalopathy, coagulopathy, varices, history of varicose bleeding, or any other clinical evidence of decompensation. (Patients with stable liver cirrhosis are eligible for this study, if a history of decompensated liver disease as outlined above has been excluded.) |
| History of alcohol or drug abuse other than cannabis within the past 12 months. Patients with documented drug and alcohol addiction free history of at least 12 months who are, in the opinion of the investigator, unlikely to relapse, may be enrolled in the study. |
| Body mass index < 18 or > 35 kg/mm2 |
| Usage of any investigational drugs within 12 months before enrolment; or the planned usage of an investigational drug during the course of the current study |
| Known hypersensitivity to any ingredient of the study drugs |
| A condition that is defined as one which in the opinion of the investigator may put the patient at risk because of participation in the study or may influence the results of the study or the patient’s ability to participate in the study. |
| Active malignancy or history of malignancy within the last 5 years (with the exception of appropriately treated basal cell carcinoma of the skin or in situ carcinoma of the uterine cervix). Suspected but yet unproven hepatocellular carcinoma. |
| Alpha fetoprotein value >100 ng/mL at screening; if > 20 ng/mL and ≤ 100 ng/mL, patients can be included if there is no evidence of liver cancer in an appropriate imaging study (e.g., ultrasound, CT scan, MRI) within past 6 months of Day 1. |
| Total bilirubin >2 x ULN with ratio of direct/indirect > 1 (Patients with Gilbert’s syndrome are not excluded.) |
| ALT or AST level >3 x ULN |
| Prothrombin time INR (Institutional Normalised Ratio) prolonged to >1.5 |
| Hemoglobin < 11.5 g/dL for women and < 12.5 g/dL for men |
| White blood cell count < 2,000 cells/mm3 |
| Absolute neutrophil count < 1,500 cells/mm3 |
| Platelet count < 90,000 cells/mm3 |
| TSH, fT3 and fT4 outside normal limits and no adequately controlled thyroid function; patients with TSH below the lower limit of normal may be enrolled if fT4 is normal and there is no clinical evidence of hyperthyroidism or hypothyroidism. |
| Poorly controlled diabetes mellitus as evidenced by HbA1c > 7.5% |
| Hemoglobinopathy (e.g., thalassemia major or sickle cell anemia) |
| History of moderate, severe or uncontrolled psychiatric disease, especially depression, including a history of hospitalisation or prior suicidal attempt; patients with a history of mild, stable depression may be enrolled provided that a pre-treatment assessment of the patient’s psychiatric disease supports that the patient is clinically stable. |
| Clinical evidence of chronic cardiac disease (e.g., coronary artery disease, congestive heart failure, uncontrolled hypertension, significant arrhythmia) |
| Clinically significant abnormalities on screening ECG |
| Clinical evidence of chronic pulmonary disease (e.g., chronic obstructive pulmonary disease) associated with functional impairment |
| Active autoimmune disease, including autoimmune hepatitis |
| Organ transplant history, other than cornea or hair |
| Active seizure disorder within the past 2 years; patients may be enrolled if on stable medication and seizures have not been experienced for more than 2 years before enrolment. |
| Requirement for chronic systemic corticosteroids (nasal or pulmonary steroids will be allowed) |
| Pregnancy and lactation |
| Medical or psychological condition that would not permit completion of the trial or signing of informed consent |
Table 2: Inclusion and exclusion criteria.
Patient consent and ethics approval: This trial protocol has been evaluated and approved by the representative ethics committee (file number 837.462.11 (8004), Landesärztekammer Rheinland-Pfalz, Mainz, Germany) at the leading study site. Ethics committees in charge at the different study sites in Aachen, Berlin, Bonn, Düsseldorf, Freiburg, Frankfurt, Hannover, Hamburg, Heidelberg, Homburg, Kiel, Köln, Leipzig, München, Regensburg, Tübingen, Ulm and Würzburg (all Germany) have approved the study protocol under the file number 837.462.11 (8004) (for details see suppl. information). Written informed consent is obtained from all trial participants prior to study inclusion. Investigators at each study site are ensuring that the study is maintained in concordance with German law as well as ethical standards following GCP regulations and the Declaration of Helsinki.
Investigational intervention
All patients are planned to continue their NUC medication with lamivudine, adefovir, entecavir or tenofovir or a combination of those during the trial until at least the end of follow-up. The treatment group additionally receives open-label peg-IFNα (Pegasys®, Roche, Germany) at a dose of 180 µg administered subcutaneously once weekly for 48 weeks. The initial injection is given in the trial center at the baseline visit. Patients are taught self-administering injections and are encouraged to self-administer all subsequent doses. Compliance is assessed by counting of empty containers and unused medication at study visits. Details of peg-IFNα administration are recorded in the electronic case report form (eCRF).
Temporary interruptions of study drug(s) administration are discouraged; and the dose of peg-IFNα should not be changed during the trial, unless dose reduction is indicated because of laboratory abnormalities or adverse events (Table 3). When a peg-IFNα dose modification is required for moderate to severe adverse reactions, a stepwise dose reduction to 135 μg or 90 μg may be applied. Following improvement of the adverse reaction, re-escalation of the peg-IFNα dose may be considered. The control group does not receive peg-IFNα as add-on treatment.
| Laboratory Values | Reduce peg-IFNα to | Discontinue peg-IFNα if |
|---|---|---|
| ANC | ≥750μl: Maintain 180 μg < 750/μl: Reduce to 135 μg |
< 500/μl: Treatment should be suspended until ANC values return to more than 1000/mm3 Re-institute at 90μg and monitor ANC |
| Platelet | ≥ 50.000/μl: Maintain 180 μg < 50.000/μl: Reduce to 90 μg |
<25.000/mm3 |
| ALT | > 10 x ULN and >2 x baseline values: Reduced to 135 μg | ALT increases are further progressive despite dose reduction or are accompanied by increased bilirubin or evidence of hepatic decompensation |
Table 3: Laboratory dose adjustments.
After the end of the trial, patients who experienced a complete anti-HBs seroconversion (i.e. who become HBsAg negative and anti-HBs positive) will stop all antiviral therapy. HBeAg negative patients without anti-HBs seroconversion will continue therapy with NUCs as previously.
Concomitant medication: The following concomitant treatments are not permitted during the trial: telbivudine, systemic corticosteroids (used for longer than 14 days) or other immunosuppressive medications (e.g. cyclosporins, tacrolimus, methotrexate etc.), systemic cytostatic chemotherapy, immuno-modulating agents such as IL-2, anti-TNF-antibodies or rituximab, nephrotoxic agents such as aminoglycoside antibiotics, amphotericin B, foscarnet, ganciclovir, non-steroidal anti-inflammatory drugs, or other agents with significant nephrotoxic potential, hepatotoxic agents such as anabolic steroids, isoniacid, itraconazole, ketoconazole, rifabutin, rifampin, or other agents with significant hepatotoxic potential. If a patient needs treatment with any excluded concomitant medication, the sponsor should be contacted prior to the initiation of the new medication.
Randomization
Patients are randomised in a 2:1 ratio (peg-IFNα in addition to NUCs vs. NUCs therapy) using block randomisation stratified by HBsAg at screening (<20.000 IU/ml vs. ≥ 20.000 IU/ml) after checking if all inclusion criteria are met and no exclusion criterion is met. The patients are not stratified for the NUC regimen applied during the trial. Randomisation lists based on a variable random block size were generated at IZKS Mainz by means of a SAS® (SAS Institute, Cary, USA) program. The randomisation lists are kept in safe and confidential custody at IZKS Mainz. A web based randomisation tool developed by IZKS Mainz is used within this trial allowing investigators to randomise patients via a secure web interface. Role specific access rights and the need to confirm all details necessary for stratified randomisation (HBsAg at screening (<20.000 IU/ml vs. ≥ 20.000 IU/ml)) are incorporated within the tool and will reduce the risk of misuse and unintended randomisations.
Withdrawal criteria and lost to follow-up
Patients can withdraw their consent at their own request without given reasons at any time during the trial. This should be without any disadvantages for the patient. However, the investigator should try to perform a final visit to get concluding findings of investigation. In addition, patients may be withdrawn from the trial for the reasons listed in Table 4. In all cases, the reason for withdrawal must be recorded in the eCRF and in the patient’s medical records. In case of withdrawal of a patient at his/her own request, if possible, the reason should be asked for and documented. The patient must be followed up and if possible, all examinations scheduled for the final trial day should be performed and documented. A patient will be considered lost to follow-up if the investigator is not able to contact him despite multiple attempts (at least 2 telephone contacts plus 1 mailing).
| Withdrawal criteria |
|---|
| If the patient withdraws his/her consent at their own request without given reasons at any time during the trial |
| If, in the investigator’s opinion, continuation of the trial would be detrimental to the patient’s well-being. |
| For women, if it becomes known that the patient is pregnant. |
| If development of a toxicity or adverse event warrants drug discontinuation. |
| If polyneuropathy develops and is confirmed by a neurologist, withdrawal will be discussed according to severity of the symptoms |
| If the patient has to take any concomitant drug interfering with the study medication |
| •If the patient needs any excluded concomitant medication as follows •Telbivudine •Systemic corticosteroids, if used for longer than 14 days, or other immunosuppressive medications (e.g. cyclosporines, tacrolimus, methotrexate etc.) •Systemic cytostatic chemotherapy •Immune-modulating agents such as IL-2, anti-TNF-antibodies or rituximab •Nephrotoxic agents such as aminoglycoside antibiotics, amphotericin B, foscarnet, ganciclovir, NSAID, or other agents with significant nephrotoxic potential •Hepatotoxic agents such as anabolic steroids, isoniacid, itraconazole, ketoconazole, rifabutin, rifampin, or other agents with significant hepatotoxic potential |
| If patient is no longer able to participate for other medical reasons (e.g., surgery, adverse events, pregnancy or other diseases). |
Table 4: Withdrawal criteria.
Physical examination
Vital signs (body temperature, heart rate and blood pressure) and weight are determined at every visit, height at screening only. Complete physical examinations are performed at screening, week 48, week 72, and early discontinuation (if applicable), symptom-directed physical examination at all other visits. Special attention should be paid to the development of polyneuropathy or other neurological symptoms in patients on combination therapy. If neurological symptoms occur, they need to be documented as adverse events, and the patients will then be referred to a neurologist for further examination.
Diagnostic procedures
All patients must undergo hepatic ultrasound or computed tomography scan or magnetic resonance imaging to rule out hepatocellular carcinoma (HCC) within 6 months before taking part in this study. A standard 12-lead electrocardiography is performed at screening visit.
Laboratory tests
Laboratory test panels are scheduled throughout the trial as outlined in Table 5a and 5b. HBV-DNA (HBV-specific polymerase chain reaction, PCR), HBsAg and anti-HBs are measured centrally at the department of clinical chemistry at Mainz University Medical Center. All other laboratory tests, including HBeAg, anti-HBe, anti-HBc, hepatitis C virus, hepatitis D virus, human immunodeficiency virus serology, routine hematology, clinical chemistry, and urine screening tests (drug screening and urinalysis) are performed locally in each study center by an accredited laboratory. Urine pregnancy test for women of childbearing potential is performed using commercially available tests at the trial sites.
| Visit 1 | Visit 2 | Visit3 | Visit4 | Visit5 | Visit6 | Visit7 | Visit8 | Visit9 | Visit 10 | Visit 11 | Visit 12 | Visit 13 | Visit 14 | Visit 15 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Screening | Baseline | EoT | EoFU | ||||||||||||
| Week -4 to 0 | Week 0 | Week 4 | Week 8 | Week 12 | Week 16 | Week 20 | Week 24 | Week 28 | Week 32 | Week 36 | Week 40 | Week 44 | Week 48 | Week 72 | |
| Informed consent | X | ||||||||||||||
| Medical history | X | ||||||||||||||
| Eligibility criteria | X | ||||||||||||||
| 12 Lead ECG | X | ||||||||||||||
| Virology | X | ||||||||||||||
| Liver assessment | X | ||||||||||||||
| Drug screening, urinalysis | X | ||||||||||||||
| Pregnancy screening | X | x | x | x | x | ||||||||||
| Randomisation | x | ||||||||||||||
| Physical examination1 | C | T | T | T | T | T | T | T | T | T | T | T | T | C | C |
| AEs2, concomitant med. | X | x | x | x | x | x | x | x | x | x | x | x | x | x | x |
| Routine laboratory tests3 | III | I | I | I | II | I | I | II | I | I | I | I | I | II | II |
| HBV-PCR | X | x | x | x | x | ||||||||||
| HBsAg / anti-HBs | X | x | x | x | x | x | x | x | x | x | x | x | x | x | x |
| Immunology4 | x | x | x | x | |||||||||||
| Bio-5 and genetic markers6 | x5,6 | x5 | x5 | x5 | x5 | x5 | x5 | x5 |
Table 5a: Trial schedule – Treatment group (peg-IFNα and nucleos(t)ide).
| Visit 1 | Visit 2 | Visit3 | Visit4 | Visit 5 | Visit 6 | Visit 7 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Screening | Baseline | EoT | EoFU | ||||||||||||
| Week -4 to 0 | Week 0 | Week 4 | Week 8 | Week 12 | Week 16 | Week 20 | Week 24 | Week 28 | Week 32 | Week 36 | Week 40 | Week 44 | Week 48 | Week 72 | |
| Informed consent | x | ||||||||||||||
| Medical history | x | ||||||||||||||
| Eligibility criteria | x | ||||||||||||||
| 12 Lead ECG | x | ||||||||||||||
| Virology | x | ||||||||||||||
| Liver assessment | x | ||||||||||||||
| Drug screening, urinalysis | x | ||||||||||||||
| Pregnancy screening | x | x | x | ||||||||||||
| Randomisation | x | ||||||||||||||
| Physical examination1 | C | T | T | T | T | C | C | ||||||||
| AEs2, concomitant med. | x | x | x | x | x | x | x | ||||||||
| Routine laboratory tests3 | III | I | II | II | I | II | II | ||||||||
| HBV-PCR | x | x | x | x | x | ||||||||||
| HBsAg / anti-HBs | x | x | x | x | x | x | x | ||||||||
| Immunology4 | x | x | |||||||||||||
| Bio-5 and genetic markers6 | x5,6 | x5 | x5 | x5 | x5 | x5 |
Table 5b: Trial schedule – Control group (nucleos(t)ide only).
Routine laboratory test panels
Laboratory panel I: Erythrocytes, hemoglobin, hematocrit, platelets, total leukocytes and differential, alanin-aminotransferase (AST), aspartat-aminotransferase (ALT), total bilirubin, gamma-glutamyltransferase (gamma-GT), alkaline phosphatase, total bilirubin (and conjugated bilirubin if total bilirubin is >1.5 × ULN), lactate dehydrogenase (LDH), sodium, potassium, calcium, creatinine (and calculated clearance), blood urea nitrogen (BUN), uric acid, glucose, prothrombine time (PT), international normalized ratio (INR)
Laboratory panel II: Includes all parameters of laboratory panel I plus albumin, thyreoid-stimulating hormone (TSH), free triiodothyronine (fT3), free tetraiodothyronine (fT4).
Laboratory panel III: Includes all parameters of laboratory panel I plus albumin, TSH, fT3, fT4, alpha-fetoprotein (AFP), immune globuline G (IgG), anti-nuclear antibody (ANA), anti-smooth muscle actin antibody (SMA), anti-soluble liver antigen antibody (SLA), anti-liver-kidney microsomal antibody (LKM), anti-mitochondrial antibody (AMA), anti-neutrophil cytoplasmatic antibody (ANCA), ferritin, lipid profile (triglyceride, total cholesterol, high density lipoprotein (HDL), low density lipoprotein (LDL)), HLA typing, urinalysis
Assessment of efficacy
Quantitative HBs-Antigen is measured every four or twelve weeks in the intervention or control arm, respectively, to address the decline of quantitative HBsAg during therapy and at the end of treatment after 48 weeks. Alternative serological markers of hepatitis B (quantitative HBsAg, anti-HBs, HBeAg, anti-HBe), HBV-DNA and liver function tests are routinely tested as scheduled. Since a genomic analysis of the virus is not possible in patients with non-detectable HBV-DNA, serotypic determination of HBV type A-D is performed. This assay shows a coincident rate with genotyping of 95-100% [8].
Assessment of safety
Patients must be carefully monitored for adverse events (AE) by the investigator at each scheduled visit. This includes monitoring of vital signs, physical exam findings, laboratory test abnormalities and changes of laboratory test values over time. The period of observation for adverse events extends from the time point of informed consent up to end of follow-up at week 72. The investigator should inform a patient when medical care is needed for intercurrent illness (ES) of which the investigator becomes aware. Adverse events are classified according to GCP definitions. The seriousness, intensity and the causal relation to trial medication and/or procedures (according to modified WHO criteria of 1991) are to be assessed by the investigator.
Serious AEs (SAEs) must be immediately (within 24 hours of the investigator’s awareness) reported to the IZKS Mainz. The investigator should provide related additional information on the clinical course and the outcome of each serious AE as soon as possible (follow-up report). SAE management and the expedited reporting follow the German Drug Law (AMG) and GCP regulation (GCP-V) is conducted by the IZKS Mainz. AEs reported by the patient or detected by the investigator are documented in the eCRF and in the patient’s medical records. Any pregnancy diagnosed in a female patient or in the female partner of a male patient during treatment with the investigational product must be reported immediately using a pregnancy report form. Relevant additional illnesses present at the time of informed consent are regarded as concomitant illnesses to be documented in the eCRF.
Trial schedule
The screening (visit 1) should take place within 28 days before baseline (visit 2). Patients should undergo a re-screening (visit 1.1) if start of treatment is delayed. Patients who have a laboratory test value outside the range specified by the inclusion criteria may have the test repeated to determine eligibility; however, the result must be available prior to visit 2 (day 1).
At screening visit informed consent, eligibility criteria and demographics (date of birth, gender, ethnic origin/nationality, height and body weight) are obtained. Medical history, concomitant medication, alcohol consumption and prior/concomitant medication for HBV (standard interferon, peg-IFNα, NUCs: start of therapy, dosage regimen (start and end dose), cessation of therapy; and vaccination for HBV) are documented. Liver assessment includes documentation of liver biopsy results (e.g. histopathological scoring by METAVIR, Ishak, Knodell or Scheuer) and documentation from non-invasive methods (e.g. liver elastography by FibroScan®) if available.
If a patient has been determined eligible by the investigator, the patient is randomised and assigned a randomisation number via a web-based randomisation tool at baseline visit (visit 2). Sites must enter the treatment allocation on the appropriate eCRF pages. If appropriate, treatment with peg-IFNα is started after randomisation. The first peg-IFNα dose should be administered under the supervision of the investigator (or a designee) at the study center.
Additional study visits are scheduled every 4 weeks in the treatment group (visits 3-14) and every 12 weeks in the control group (visit 3-7). After the end of treatment (week 48) all patients are followed for additional 6 months until the end of follow-up visit. Throughout all study visits physical examination, acquisition of laboratory tests and blood samples are scheduled as indicted in Table 5a and 5b.
Statistics
Details of the statistical analysis of the data collected in this trial will be documented in a statistical analysis plan (SAP) that will be finalized before closing the data base. The SAP is based on the protocol including all amendments. The document may modify the plans outlined in this protocol; however any major modifications of the primary endpoint definition and/or its analysis will also be reflected in a protocol amendment. Any deviation from the original statistical plan must be described and justified in the final report. The statistical analysis will be conducted by means of SAS®.
Sample size calculation
Based on pilot data, it is assumed that 12% of the patients in the peg-IFNα/NUC group HBsAg will drop 1log10 after 48 weeks of combination therapy, while only 0.1% of the patients without additional interferon will reach this endpoint. Studies in hepatitis C patients reported of a discontinuation rate due to tolerability and safety of approximately 10-15% [9]. As this rate was observed in combination with ribavirin we assume a drop-out rate of 10% due to better tolerability and safety in hepatitis B patients.
The assumed difference in HBsAg loss rates will be detected with a power of at least 80% by Fisher's exact test at a two-sided significance level of 5%, if 153 patients (102 in the interferon arm, 51 in the control arm) can be analysed. Taken into account a drop-out rate of 10%, 170 patients will be randomised. Patients with no HBsAg measurement at week 48 will be analysed by their last HBsAg measurement (last observation carried forward). This approach is considered slightly conservative but allows all randomised patients to be included into the confirmatory modified Intention to treat analysis.
Analysis populations
All patients who signed informed consent were assigned a randomisation number and are considered as enrolled/randomised patients, even if they did not receive any trial treatment. All patients who received at least one dose of trial treatment and with at least one available post-baseline assessment of the primary analysis variable will be included in the modified Intention-to-treat (mITT) population. Therefore the mITT population, which represents the primary analysis population, may not include all randomized patients. Within mITT population analyses patients will be assigned to the treatment to which they were randomised.
To be eligible for the per protocol population, patients must receive the trial treatment as randomized with a treatment compliance of at least 90%. In addition eligible patients may not violate inclusion criteria, meet any exclusion criteria, receive not permitted concomitant treatment and deviate from planned visit schedules. The safety population comprises all patients who received at least one dose of trial treatment. In analyses of the safety population patients will be assigned to the treatment, which they actually received.
Efficacy analyses
The primary population for the analyses of efficacy is the mITT population. All hypotheses will be tested on a two-sided level of significance α=0.05. The primary endpoint is assessed by the null hypothesis: HBsAgRRInterferon=HBsAgRRControl and the corresponding alternative hypothesis: HBsAgRRInterferon ≠ HBsAgRRControl. (HBsAgRR denotes HBsAg response rates, ≥ 1log10 drop of HBsAg at week 48 compared to baseline. The subscripts show the respective treatment group.)
The response rates will be compared between the treatment groups by Fisher’s exact test. In the primary analysis patients with missing HBsAg measurements at week 48 will be analysed according to the last observation carried forward principle thus all patients in the modified Intention to treat population are available for analysis. As needed, alternative methods such as multiple imputation or repeated measures mixed models will be applied as sensitivity analyses. The primary analysis (Fisher’s exact test) will be repeated for the per protocol population. Furthermore, a logistic regression model including covariates assumed to predict treatment response (HBV genotype, age, sex, duration of antiviral therapy before the study) will be applied.
Analysis of secondary endpoints and subgroup analysis
The primary analysis population for the secondary endpoints is the mITT population. Secondary endpoints will be analysed using appropriate methods depending on the scale of the respective parameter. Analyses of secondary endpoints will be interpreted descriptively. Age, sex, HBV genotype, level of quantitative HBsAg, NUC regimen (substances, combination, duration) will be subject of subgroup analyses.
Interim analyses
There will be no interim analysis in the sense that the primary endpoint is analysed prematurely and a decision about the continuation of the study is derived. However, it is planned to compare the rates of patients with at least 10% HBsAg loss 24 weeks after the start of study treatment compared to baseline between the treatment groups. This interim analysis is performed once as soon as sufficient 24 week follow up data are available. The analysis will be interpreted in an explorative manner. Therefore no adjustment of the significance level is required.
Analysis of adverse events
All summaries and listings of safety data will be performed for the safety population. Frequencies of patients experiencing at least one adverse event (AE) will be displayed by system organ class and preferred term according to MedDRA terminology. A patient listing of all AEs will be prepared. Detailed information collected for each AE will include: A description of the event, duration, whether the AE was serious, intensity, relationship to trial drug, action taken, and clinical outcome. Summary tables will present the number of patients observed with AEs and corresponding percentages. Additional subcategories will be based on event intensity and relationship to trial drug.
Analysis of clinical and laboratory findings
Listings will be prepared for each laboratory measure and will be structured to permit review of the data per patient as they progress on treatment. Summary tables will be prepared to examine the changes of laboratory measures over time. Additionally, shift tables will be provided to examine the changes of laboratory data from normal baseline to values outside the corresponding reference range during/after treatment. Changes in laboratory parameters are reported as adverse events if the test result is associated with accompanying symptoms, and/or requires additional diagnostic testing or medical/surgical intervention, and/or leads to a change in trial dosing outside of protocol-stipulated dose adjustments. Further changes in laboratory parameters are reported as adverse events if they lead to discontinuation from the trial, significant additional concomitant drug treatment, or other therapy, and/or are considered to be an adverse event by the investigator or sponsor.
Changes in vital signs (blood pressure, heart rate and temperature) and, cardiologic evaluation by electro cardiogram (ECG) will be analysed descriptively by distributional parameters (such as mean and standard deviation) or absolute and relative frequencies.
The PADD-ON trial aims to evaluate the peg-IFNα add-on treatment as an approach to improve immune control in cHB, which could eventually allow a complete halt of antiviral treatment. HBeAg negative cHB patients under stable HBV suppression were selected for the PADD-ON trial, as this represents the largest subgroup all cHB patients receiving NUC therapy in Europe and North America [10]. In addition no scenario for the cessation of NUC treatment has been defined for HBeAg negative cHB beside anti-HBs seroconversion, which is a very rare event during NUC therapy. Inclusion of HBeAg negative cHB patients was further limited to patients with serum HBsAg ≥100 IU/mL to avoid a selection bias for patients with favourable treatment outcome or spontaneous HBsAg clearance prior to study inclusion.
The peg-IFNα treatment schedule for 48 weeks applied during the PADD-ON trial is based on the standard of care for cHB patients, which allows prediction of safety issues related to peg-IFNα. However, the treatment duration of 48 weeks might not be sufficient to eliminate HBsAg from the circulation in all patients, showing an initial response. The PADD-ON trial therefore addresses the dynamics of HBsAg reduction as the primary endpoint, which reflects increasing immunological control of HBV transcription. Complete HBsAg clearance and anti-HBsAg seroconversion were included as secondary endpoints. The robust laboratory primary and secondary endpoint selection further reduces bias by the open-label trial design due to the subcutaneous peg-IFNα administration route precluding any blinding.
Following the EOT the PADD-ON protocol provides a follow-up period, to address delayed IFN effects on immune control, as observed for anti-HBe seroconversion and HBsAg decline post IFN administration [11,12]. Patients who developed an anti-HBs seroconversion until EOF will be able to stop antiviral therapy, which is in line with current treatment guidelines. Patients showing no HBsAg response, HBsAg decline or HBsAg elimination at EOF will undergo standard of care treatment (NUC, peg-IFNα or surveillance) according to the investigators decision. The variable treatment options after EOF, should therefore prone a long-term observational study to characterize the adequate management after the peg-IFNα add-on intervention.
MS, JK, JS and PG received lecture fees from Roche Pharma AG (Grenzach-Wyhlen, Germany). UA is employee of Roche Pharma AG (Grenzach-Wyhlen, Germany). The other authors declare that they have no competing interests. The legal sponsor of this trial is the University Medical Centre of the Johannes Gutenberg University Mainz. The First Medical Department (University Medical Center, Mainz) executing the PADD-On trial, receives financial support from Roche Pharma AG (Grenzach-Wyhlen, Germany).
MFS and AG made substantial contributions to conception/ design of the trial protocol and drafted the manuscript. JK, JS, MW, MS and PG contributed to the conception/ design of the trial protocol and revised the manuscript. DW, AE and CR provided the statistical analysis plan and revised the manuscript. All authors read and approved the final manuscript.
We would like to acknowledge Susan Depoix, Ulrike Belzer-Endres and Brigitte Bartsch for excellent management of laboratory data, trial correspondence and technical assistance. We acknowledge IZKS Mainz (funding numbers FKN 01KN0703 and FKN 01KN1103, IZKS Mainz of the German Federal Ministry of Education and Research) for conducting the sponsor function: study coordination, regulatory support, clinical monitoring, data management, safety management and statistics.