Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Short Communication - (2015) Volume 3, Issue 5
Iron deficiency anemia (IDA) is the commonest type of anemia all over the world. It is easily treatable with oral iron therapy, but a minority of patients does remain unresponsive. This may be related to inappropriate compliance, manifest or occult blood loss, worm infestations, helicobacter infection, known malabsorption syndromes or paroxysmal nocturnal hemoglobinuria (PNH). We observed six such patients with non-responsive recurrent classical IDA who did not show any of the above conditions on specific investigations.
Iron tolerance test (as an index of oral iron absorption) was performed on these patients. All of them showed negligible or no iron absorption, suggesting a defect in intestinal oral iron absorption as the cause of non-responsive IDA. This contention was supported by a dramatic response to parenteral iron therapy in all of them. Since the tests for known malabsorption syndromes were negative, it would be assumed that these subjects had an isolated defect in oral iron absorption that has been only scantily reported in the literature. Five of the six patients were siblings of two families. This raises the possibility of the defect in iron absorption to have a genetic connotation. Pending the complete work up for genetic studies, including the gene mutation for membrane bound serum protease type 6 (TMPRSS6) in these subjects and their families, this presentation is a short communication to expose and share our small experience of what looks to be an isolated defect in intestinal iron absorption.
Keywords: Non responsive iron deficiency anemia; Iron absorption; Iron tolerance test; Defective oral iron absorption
Iron deficiency anemia (IDA) is believed to be the commonest type of anemia occurring all over the world, including United States of America [1]. Most commonly, it is caused by nutritional iron deficiency and the anemia is easily corrected with adequate supply of food or medicinal iron. However, a minority of IDA patients remain unresponsive. Nearly 50 cases have been described in medical literature [2,3]. For the cause of non-responsive IDA (NR-IDA) in such patients, attention is ordinarily drawn to noncompliance, occult blood loss, worm infestations, helicobacter infection, known malabsorption syndromes and paroxysmal nocturnal hemoglobinuria (PNH).
We have observed six patients with recurrent NR-IDA in whom all these causes were excluded. And all of them recovered dramatically, with intravenous (IV) iron therapy, suggesting defective intestinal iron absorption as the cause of non-responsiveness to oral iron therapy. This was supported by the results of iron tolerance test [4] that we had standardized as an index of iron absorption in our local population. It showed unequivocal evidence of almost complete lack of iron absorption in all our six patients. Moreover, all the six patients showed dramatic response to IV iron administration with complete correction of anemia, providing further assurance that defective iron absorption was the sole reason for their IDA. In view of the absence of the evidence of any known malabsorption syndrome, the possibility of an isolated defect in intestinal iron absorption was considered as the most likely causative factor in our patients. Three of these patients (aged 17, 19 and 23) were siblings of one family and two patients (aged 7 and 15) were siblings of another family, suggesting the possibility of a genetic mechanism for their defective iron absorption. The sixth patient was the only child in the family.
We are still working on the genetic studies in these patients and their families. In the meanwhile, this presentation is a preliminary short communication to attract information on experience from other workers.
The present study was performed prospectively on six patients who had reported with history of recurrent anemia and had shown no response to oral medicinal treatment including adequate iron therapy.
The relevant clinical profile of these patients is shown in Table 1 and the results of preliminary laboratory investigations are shown in Table 2.
| Name | Age and sex | Date included in study | Duration of illness | Symptoms of anemia | Koilonychias | Liver | Spleen |
| MAR | 17F | Oct. 2010 | 2 years | present | present | Normal | Normal |
| TRR | 19F | Oct. 2010 | 2 years | Present | Present | Normal | Normal |
| GMR | 23F | Oct. 2010 | 3 years | Present | Present | Normal | Normal |
| LMT | 13F | Feb. 2011 | 1 year | present | None | Normal | Normal |
| SLT | 7F | Feb.2011 | 8 months | Present | None | Normal | Normal |
| GA | 11M | Aug.2012 | 2 years | Present | None | Normal | Normal |
Table 1: The relevant clinical findings in 6 patients of the present study.
| Name of the patient | MAR | TRR | GMR | LMT | SLT | GA |
| HB ((g/L) | 6.8 | 8.1 | 7.3 | 7.1 | 6 | 7.3 |
| MCV (fl) | 69.9 | 72.8 | 71.9 | 76.3 | 69.7 | 72.6 |
| MCH (pg) | 19.6 | 20.7 | 19.8 | 20.1 | 18.7 | 23.2 |
| Blood smear findings | POS | POS | POS | POS | POS | POS |
| Serum iron (umol/L) | 3.2 | 5.7 | 6.1 | 5.3 | 4.7 | 4.9 |
| Serum transferrin (g/L) | 3.9 | 3.1 | 4.2 | 3.8 | 3.7 | 4.2 |
| Serum ferritin (ng/ml) | 4.7 | 6.8 | 4.9 | 5.8 | 7.2 | 6.4 |
| HB electrophoresis | ||||||
| Hb A (%) | 98.8 | 97.2 | 96.9 | 98.2 | 97.1 | 97.3 |
| Hb A2 (%) | 1.2 | 2.8 | 3.1 | 1.8 | 2.9 | 2.7 |
| Bone marrow | ||||||
| Giemsa stain | MN | MN | MN | MN | MN | MN |
| Prussian Blue stain for iron | DI | DI | DI | DI | DI | DI |
Table 2: The results of relevant laboratory investigations in the six patients of the present study (at the time of recruitment in the study). POS: Positive for Iron Deficiency Anemia (IDA) that is Microcytosis, Hypochromia, Pencil-shaped cells. No other red cell shape abnormality or evidence of increased erythropoietic reactivity/hemolytic activity. MN: Micronormoblastic erythropoiesis with dominance of miconormoblasts with reduced hemoglobinization (disproportionate nuclear maturation relative to hemoglobinization) otherwise normal. DI: Depleted hemosiderin iron (particulate, macrophagic and sideroblastic). HB: Hemoglobin; MCV: Mean Corpuscular Volume;MCH: Mean Corpuscular Hemoglobin; HbA: Haemoglobin A fraction; Hb A2: Hemoglobin A2 fraction on electrophoresis.
Symptoms of anemia
Pallor, Generalized weakness, easy fatigue, exertional dyspnoae
None of the adult female patients had history of menorrhagia, none had piles or history of bleeding or malena
Duration of illness: Duration of illness indicates the time between the patient's first consultation with the medical internist and reporting in our clinic, though the parents believed that the patients had been showing pallor since childhood.
MCH: Mean Corpuscular Hemoglobin; HbA: Haemoglobin A fraction; Hb A2: Hemoglobin A2 fraction on electrophoresis.
Of the six patients, 5 were females including two children and one male child. Three patients (MAR, TRR, GMR) were siblings of one family and 2 patients (LMT, SLT) of another family while the sixth patient (GA) was the sole child of his parents.
From the preliminary investigations that included complete blood count (CBC), peripheral blood smear examination (Giemsa stain), serum iron (immunoassay on Cobas 6000, Roche Diagnostics USA), serum transferrin (immunoassay on Cobas 6000, Roche Diagnostics USA), serum ferritin (immunoturbidimetric assay on Cobas 6000, Roche Diagnostics USA ) and HB electrophoresis (agarose gel electrophoresis on Sebia Hdrasys, USA ) all the patients were diagnosed to have classical IDA that was also confirmed with examination of bone marrow aspiration smears and trephine biopsy sections (Perl’s Prussian blue staining) showing complete depletion of hemosiderin iron (grade 0). All these patients were treated with oral iron therapy (Ferrous sulphate: 200 mg three times/day) under supervision for assured compliance. The results of response to oral iron therapy are shown in Figure 1.
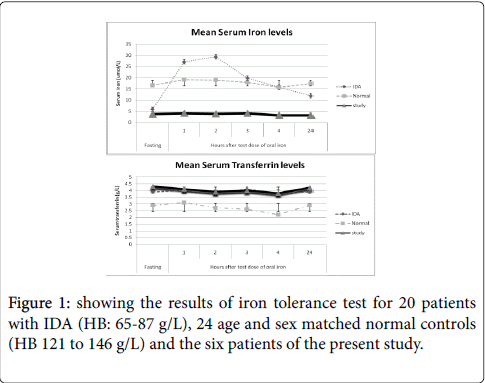
Figure 1: showing the results of iron tolerance test for 20 patients with IDA (HB: 65-87 g/L), 24 age and sex matched normal controls (HB 121 to 146 g/L) and the six patients of the present study.
None of the patients showed more than negligible response. The investigations, CBC and iron profile (serum iron, serum transferrin and serum ferritin) were also performed on the parents of all patients and the other seemingly clinically unaffected 4 siblings of the family of MAR, TRR and GMR and 3 siblings of the family of LMT and SLT. They were all found to show normal findings and had no evidence of anemia or iron deficiency.
Since these patients, in spite of having IDA did not show any response to oral iron therapy, they were investigated specifically for GI blood loss (stools for occult blood), upper GI endoscopy and colonoscopy, worm infestation (stools examination by concentration method), helicobacter infection ( endoscopic gastric biopsy and urease breath test) , malabsorption syndromes (Barium meal follow through, D-xylose test, jejunal biopsy and serology for celiac disease) and for PNH (urine for hemosiderin and Flow cytometry for CD55, CD59). The results are shown in Table 3.
| MAR | TRR | GMR | LMT | SLT | GA | |
| Stools - occult blood (x3) | NEG | NEG | NEG | NEG | NEG | NEG |
| Stools - parasite infestation | NONE | NONE | NONE | NONE | NONE | NONE |
| Upper GI endoscopy | N | N | N | N | N | N |
| Jejunal biopsy | N | N | N | N | N | N |
| Colonoscopy | N | N | N | N | N | N |
| D-xylose test | N | N | N | N | N | N |
| Barium meal follow through | N | N | N | N | N | N |
| Serum total proteins (g/L) | 77.5 | 72.8 | 69.9 | 71.8 | 70.2 | 70.8 |
| Serum albumin (g/L) | 31.7 | 31.8 | 33.2 | 29.9 | 31.4 | 30.8 |
| Serum calcium (mol/L) | 9.8 | 9.3 | 9.9 | 10.4 | 10.1 | 9.9 |
| Prothrombin time(PT)sec. | N | N | N | N | N | N |
| Activated thromboplatin time (APTT) sec. | N | N | N | N | N | N |
| Urine for hemosiderin (x3) | NEG | NEG | NEG | NEG | NEG | NEG |
| CD 55 | 96% | 93% | 97% | 94% | 96% | 93% |
| CD59 | 93% | 97% | 92% | 91% | 98% | 91% |
Table 3: The results of specific investigations performed to ascertain the cause of failure of response to oral iron therapy. POS: Positive for IDA that is Microcytosis, Hypochromia, Pencil-shaped cells. No other red cell shape abnormality or evidence of increased erythropoietic reactivity/hemolytic activity. NEG: Negative; N: Normal; GI: Gastro Intestinal; CD: Cluster of Designation.
None of the patients showed a positive finding to suggest any of the above mentioned conditions
It was, therefore, suspected that our patients suffered from some defect in intestinal iron absorption. To ascertain this, they were subjected to low dose iron tolerance test [3-5] with 10 mg of oral ferrous sulphate (Fer-in-sol in 30 ml of de-ionized iron free water), given on empty stomach followed by one hourly estimation of serum iron for 4 hours and then after 24 hours. The results are shown in Figure 2.
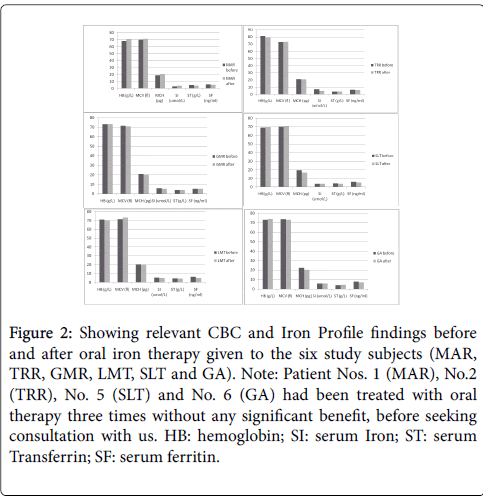
Figure 2: Showing relevant CBC and Iron Profile findings before and after oral iron therapy given to the six study subjects (MAR, TRR, GMR, LMT, SLT and GA). Note: Patient Nos. 1 (MAR), No.2 (TRR), No. 5 (SLT) and No. 6 (GA) had been treated with oral therapy three times without any significant benefit, before seeking consultation with us. HB: hemoglobin; SI: serum Iron; ST: serum Transferrin; SF: serum ferritin.
It also shows the results of our one of the previous studies that was performed to standardize and update the iron tolerance test as index of oral iron absorption in respect of our local population in 20 patients with proven and untreated IDA and 24 age and sex matched nonanemic normal controls. The iron tolerance test showed complete lack of iron absorption in each of our six patients of NR-IDA. This was followed by administration of intravenous iron therapy with ferric hydroxide sacharate complex (Ferrosac)) calculated according to the manufacturer’s (SPIMACO of Saudi Arabia) formula:
Dose of IV iron (mg)=[body weight (Kg) x HB deficit (g/L) x 0.24] + 500 mg (for storage iron)
(HB deficit=Target HB (g/L) minus existing HB (g/L)
[The factor 0.24 is derived from the following assumptions
Blood volume=approx. 7% of body weight.
Iron content of hemoglobin=0.34%
Factor 0.24=0.0034 x 0.07 x 1000 (for conversion of g to mg)].
The calculated amount of ferrosac was given in divided dose: 200 mg (100 mg for the patient, GA) dissolved in 150-200 ml normal saline on alternate days.
HB, serum iron, serum transferrin and serum ferritin were repeated at the end of every week for 20 weeks. Bone marrow aspiration and trephine biopsy was also studied for hemosiderin iron (Perl’s Prussian blue staining), after the second week following IV iron administration.
The clinical narrative of the six patients of the present study (Table 1) shows that on first presentation in our clinic, all the six patients had symptoms of chronic anemia (pallor, dizziness, easy fatigue and exertional dyspnoea). Three of them (siblings of the same family) showed koilonychia. None had dysphagia. No jaundice, hepatomegaly or splenomegaly was discovered in any patient. Although, the precise data about these patients for the period before they sought consultation in our clinic is not available, yet from interrogation and information obtained from their family physicians and treating doctors, we are confident to believe that all these patients suffered from chronic recurrent symptomatic anemia that did not respond to oral medications including high dose iron therapy since at least for the durations shown in Table 1, although the parents of 5 patients (MAR, TRR, GMR, LMT, SLT) of two families claim that the patients had been anemic since childhood.
The investigative profile of these six patients (Table 2), showed them to have classical findings of iron deficiency anemia - microcytic hypochromic picture with low serum iron, raised serum transferrin, low serum ferritin, depleted bone marrow hemosiderin iron stores and normal pattern of hemoglobin electrophoresis. They already had the history of chronic anemia that had not responded to previous oral medicinal treatments including therapeutic iron. To reassess that their anemia was non-responsive to iron therapy, they were administered oral iron therapy (ferrous sulphate 200 mg equivalent to 60 mg elemental iron, three times daily), under supervision to assure compliance. None of them showed any significant increment in HB levels or improvement in iron profile (Figure 1), reassuring thereby that all the six patients had non-responsive IDA. On specific investigations to ascertain the reason for the lack of response of IDA to assured oral iron therapy, none of them showed any evidence of occult blood loss, parasitic infestation, gastric, intestinal or colonic disease, helicobacter infection, malabsorption syndrome or PNH (Table 3). What was left to be examined was the possibility of defective gastrointestinal iron absorption. Contemporary to that time, we were coincidentally engaged in redefining and standardizing the iron tolerance test in respect of our local population by using low dose oral iron as the test dose. The results that are depicted in Figure 2, showed remarkable increments in the mean serum iron levels in IDA patients as 4.5, 4.9, 3.3, 2.6, 1.9 times of the base serum iron levels after 1 hour, 2 hours, 3 hours, 4 hours and 24 hours , after the test dose respectively. On the other hand, no significant increment (p<0.001) of serum iron levels occurred in any of our six NR-IDA study patients. This suggested a strong possibility of significant defect in intestinal absorption of oral iron as the cause of Non-responsiveness to oral iron therapy in our IDA patients (study subjects). This contention was further confirmed by the observation that all these NR-IDA patients showed dramatic response to IV iron therapy with increment of HB levels by 1-1.8 g/ week and recovery to normal values in 3-4 weeks time with serum iron levels rising to 4 to 6 times of the base level within the first week and sustaining for several weeks, as shown in Figure 3. Serum ferritin values too, although slower than the serum iron levels to rise, showed significant increments over the same period of time.
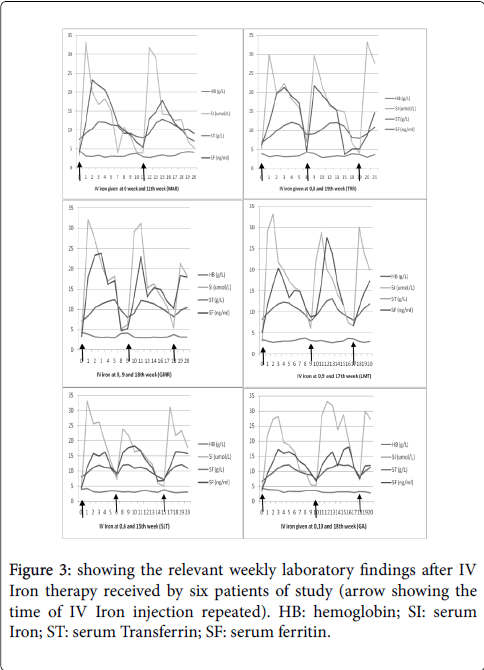
Figure 3: showing the relevant weekly laboratory findings after IV Iron therapy received by six patients of study (arrow showing the time of IV Iron injection repeated). HB: hemoglobin; SI: serum Iron; ST: serum Transferrin; SF: serum ferritin.
Bone marrow aspiration and trephine biopsy was performed on all these patients after 2 weeks onwards. The hemosiderin iron that was completely depleted before IV iron therapy, showed increment to grade 2+ to 4+ after 2-5 weeks after IV iron administration, signifying remarkable response in all these 6 patients who had been completely non-responsive to oral iron therapy. That the lack of response to oral iron therapy in our study subjects was related to lack of intestinal iron absorption is further supported by the observation that almost a similar response to IV iron therapy (Figure 3) was elicited every time when IV iron therapy was repeated after the benefit of the previously administered IV iron had weaned off in 8-11 weeks, resulting in recurrence of IDA. The fact that three of our patients were siblings of one family and 2 were siblings of another family, raises strong possibility of hereditary (genetic) connotation, on which we are still working.
The present narrative is a preliminary short communication of six patients with what we confirmed as an instance of isolated defect in gastro-intestinal iron absorption that is only scarcely described in the literature [6]. The objective is to expose our experience and to gain information on the subject from experience of others, the emphasis being on the “isolated defect” in iron absorption.
Defective iron absorption occurring as a component of general malabsorption syndrome like celiac disease or tropical sprue wherein more than one nutritional element is affected is well known. But, defect in absorption solely of iron occurring as an isolated event that is unequivocally evident in the present study, is not well described.
Observation of only 6 patients with isolated defective iron absorption over a period of 4 years may not be impressive, but five of the six patients being siblings of two families, raises an important connotation of the possibility of genetic basis for this condition. The observatioin that duration of evidence based anemia in our subject is short (Table 1), may argue against the genetic proposition as the cause of defective iron absorption in our cases, in preference to some environmental factors, bacterial infections or otherwise, that could be operative in the sibling in the same household. But firstly, the other siblings in their families (4 in one family and 3 in the other) and their parents who would have been exposed to the same environmental factors did not suffer from any degree iron deficiency or anemia. Secondly the same environmental factors to be operative in different unrelated families and that too in a few selected siblings would be a unlikely probability. Moreover it is well known that many genetic mutations may show phenotypic manifestations at a later age. In any case, the issue can be resolved only after the genetic studies in our patients and their parents have been completed.
For the sake of discussion, however, a variety of gene mutations in the membrane bound serine protease type 6 (TMPRSS6) has been described in patients with iron refractory iron deficiency anemia (IRIDA) who are non responsive to oral iron therapy due to lack of intestinal iron absorption [6-9]. With the finding of increased hepcidin levels in association with this gene mutation, it is believed that the gene mutation up-regulates the production of hepcidin that is required to sense iron deficiency [7], resulting in decreased iron absorption on the one hand and reduced release of storage iron from the macrophage pool for recycling, on the other hand [7,9]. Devoid of iron supply from both the intestinal as well as the macrophage source , the serum iron is markedly reduced (usually <2.5 umol/L) but serum ferritin is found to be inappropriately increased [10]. Therefore, the response to IV iron therapy in these NR-IDA patients is reported to be rather slow and mild [9].
Since we have not yet completed the genetic studies in our subjects, we are not ready with the precise findings about any genetic mutations in our subjects and their families, but we feel that our patients if they do have a gene mutation, it is likely to be a different mutation or a distinctly wide variant of TMPRSS6 mutation, because all the six patients in our study, had remarkably low serum ferritin levels and complete depletion of bone marrow macrophage hemosiderin iron that argues against the enhanced influence of hepcidin on macrophage iron.
The precise level at which the defect of iron absorption is operative in our patients, that needs to be worked out, is a matter of speculation at this stage. Any of the known mechanisms [11] that is conversion of trivalent ferric food iron to ferrous form , its entry into the enterocyte across the luminal side of the cell membrane via specific transporters [12], intracellular iron transfer to apotransferrin to form iron bound transferrin through specific iron channels, called ferroportins facilitated by the copper dependent protein –hephaestin (with ferroxidase activity) may be involved. In addition, the role of the iron regulating hepatic protein- Hepcidin that has inverse relationship with iron absorption needs to be evaluated in our subjects.
If the existence of isolated defect of intestinal iron absorption accompanied by complete depletion of hemosiderin storage iron (the latter being contrary to the heightened hepcidin effect), is acceptable, a lot more needs to be done not only about the site of abnormality at the gastro-intestinal level, but also at the genetic level as suggested by the defect occurring in siblings in our study