Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Research Article - (2015) Volume 3, Issue 5
Background: In females, one X-chromosome is transcriptionally silenced by methylation of CpG sites in every cell during early embryogenesis. The methyl groups needed for DNA methylation are provided by the methylenetetrahydrofolate reductase (MTHFR). A polymorphism in the enzyme (677C>T, rs1801133) affects MTHFR concentrations and possibly also methylation of X-chromosomal genes. Whether X-chromosome inactivation (XCI) by methylation is associated with the MTHFR 677C>T genotype is currently unknown. It is also unclear whether the imprinting subsequently influences factor VIII activity in carrier females.
Methods: We determined XCI in 61 hemophilia A carrier females and 174 control females by analysis of the human androgen receptor locus. Genotyping of MTHFR 677C>T was performed by mutagenically separated PCR, FVIII:C was tested by one-stage coagulation assays.
Results: Slight skewing of XCI (X1:X2<1:3) was not associated with FVIII:C activity in hemophilia A carriers and controls. In contrast, strongly skewed XCI, which was observed in 10 carrier females, was associated with FVIII:C activity levels. If the X-chromosome carrying the mutated F8 gene was methylated, FVIII:C levels were higher. In contrast, FVIII:C levels were lower if the X-chromosome with the intact F8 gene was imprinted. We found a significant association of the 677TT MTHFR genotype with low FVIII in carrier females. The MTHFR genotype was not associated with FVIII:C in control females.
Conclusion: Highly skewed imprinting of the X-chromosome influenced FVIII:C levels in carriers of hemophilia A. We found a significant association of MTHFR 677TT with low FVIII: C in hemophilia A carriers. Surprisingly, the MTHFR 677TT genotype was not significantly associated with X-chromosome inactivation. To elucidate the underlying mechanisms further studies will be necessary.
Keywords: FVIII activity; Hemophilia A carriers; Methylation; MTHFR polymorphism; X-chromosome inactivation
It is well known that plasma factor VIII activity (FVIII:C) levels are highly variable and are affected by genetic and environmental factors. In twin studies [1,2] a high degree of heritability (57% [1] and 61% [2], respectively) for FVIII was reported. In a large epidemiologic study of Spanish families [3] as well as in female relatives of hemophilia A patients [4] FVIII:C was also reported to be highly variable.
In plasma, circulating FVIII is bound to von Willebrand factor (VWF). VWF levels and polymorphisms in the VWF gene are known to have a major impact on FVIII:C [4-10]. In addition, AB0 blood groups influence FVIII:C. Individuals with blood group 0 have significantly lower FVIII:C than those with blood group A, B or AB [1,4-11]. Genes involved in FVIII synthesis and degradation have also been reported to be associated with FVIII activity [8,11-15]. Moreover, factors like age, sex, hormone levels and inflammatory processes have been found to contribute to the high variability of FVIII:C [1,3,4,10,11,16-18]. However, all these factors do not fully explain the variability of FVIII, and it is likely that other mechanisms play a role.
In females, one X-chromosome is transcriptionally silenced by methylation of CpG sites in every cell during early embryonic development [19]. Thus, females are mosaics for two cell populations – one with an inactive maternal, the other with an imprinted paternal Xchromosome. Skewed X-chromosome inactivation (XCI) patterns have been observed to be responsible for phenotypic expression of Xchromosomal diseases in females. Yoshioka et al. [20] described skewed XCI patterns in four families with Duchenne muscular dystrophy (DMD). In three symptomatic carriers of DMD, a skewed XCI of more than 70% was identified which left preferentially the mutated allele active [20]. In another study, the clinical phenotype of monozygotic twins with Rett syndrome was reported to be influenced by differences in XCI [21]. A more severe clinical manifestation of disease was found in the twin with weaker inactivation of the mutated allele. The clinical relevance of skewed XCI for the phenotypic expression of X-linked disorders like hemophilia A and B was published 2009 [22]. In this paper it was discussed that FVIII gene expression could be influenced by XCI.
Hemophilia A is caused by a variety of mutations in the factor VIII (F8) gene. It is inherited in an X-linked fashion, and thus, in most families only males are clinically affected while in female carriers the intact F8 gene compensates for the affected gene. FVIII:C varies over a wide range in hemophilia A carriers [1,23]. Occasionally, females with severe bleeding complications are reported. Some studies have shown that FVIII deficiency in females can be caused by complete skewing of XCI [24-29]. To distinguish between the active and the inactive Xchromosome and to determine the inactivation status of the Xchromosomes the HUMARA (human androgen receptor) assay [30,31] has proven useful. Highly polymorphic CAG-repeats in the HUMARA gene in combination with cleavage by methylation-specific restriction enzymes allow accurate and reproducible analysis of XCI in approximately 90% of females [30,32].
DNA methylation is regulated by two specific enzymes: the methionine synthase and the methylenetetrahydrofolate reductase (MTHFR). Both enzymes are involved in the de novo synthesis of methyl groups [33]. MTHFR, an important enzyme in folate metabolism and DNA synthesis, is responsible for the irreversible conversion of 5,10-methylenetetrahdyrofolate to 5-methylhydrofolate which is needed for the generation of methionine, the major methyl donor for DNA and histone transferases [34]. The MTHFR activity is influenced by the common MTHFR 677C>T polymorphism (rs1801133, g.14783 C>T, p.Ala222Val) [34]. The 677T variant induces a reduced enzyme activity, and individuals with MTHFR 677CT have a mean MTHFR activity of 65%, carriers of the MTHFR 677TT genotype have only approximately 30% enzyme activity [35].
In the present study, we analyzed the influence of XCI on FVIII activity in healthy females and proven carriers of hemophilia A. Furthermore, we evaluated the association of the MTHFR 677C>T genotype with X-chromosome inactivation in blood cells, and we determined a correlation between the MTHFR 677C>T genotype and FVIII:C.
Study population
Sixty-one female hemophilia A carriers of 46 different families were recruited between 2006 and 2011 at the hemophilia care centers of the Department of Medicine I, Clinical Division of Hematology and Hemostaseology and the Department of Pediatrics at the Medical University of Vienna and at the Department of Internal Medicine I at the hospital of Klagenfurt am Woerthersee. After written informed consent was obtained from all study participants the MTHFR genotype, the degree of XCI and FVIII activity were determined. In a subgroup of hemophilia A carriers (n=26) we could identify which Xchromosome carried the mutated F8 gene. Females in group A (n=13) carried the intact F8 gene on the inactivated, methylated Xchromosome, individuals in group B (n=13) had their intact F8 gene on the active, unmethylated X-chromosome.
The control population comprised 174 healthy females from the same ethnic background and geographic region as the carriers. Controls were either spouses or friends of patients treated at the Clinical Division of Hematology and Hemostaseology or acquaintances or relatives of hospital staff. All probands were interviewed regarding their history of bleeding tendency and thrombotic events. They were not related to each other nor related to the hemophilia A carriers.
The study was approved by the Ethics Committee of the Medical University of Vienna.
Methods
Blood sampling and laboratory analysis: Blood samples were drawn into Vacuette vacuum tubes with sodium citrate or EDTA as anticoagulants (Greiner-Bio One, Kremsmuenster, Austria) for clotting and genetic analysis, blood chemistry and blood count measurement and the determination of AB0 blood groups. FVIII:C levels were determined by a one-stage coagulation assay on a Sysmex CA-7000 analyzer (Sysmex Corporation, Kobe, Japan) using FVIII-deficient plasma (Technoclone, Vienna, Austria) and Dade Actin FS activated PTT reagent (Dade Behring, Marburg, Germany). Von Willebrand factor antigen (VWF:Ag) was assayed on a STA-R analyzer (Diagnostica Stago) using the STA LIATest VWF immunoassay (Diagnostica Stago).
DNA extraction and mutation analysis: DNA was isolated from peripheral blood using the MagNA Pure LC DNA isolation system (Roche Diagnostics, Mannheim, Germany) according to the manufacturer’s instructions.
In all carriers of hemophilia the presence of a mutation in the F8 gene was confirmed by analyses of inversion 1 and 22 as well as sequence analyses. The inversion in intron 22 of the F8 gene was tested using the method described by Liu et al. [36]. The analysis of the inversion in intron 1 was carried out using a slightly modified protocol of Bagnall et al. [37]. In carriers with no inversion F8 gene sequencing was performed using the Big Dye Terminator v3.1 cycle sequencing kit (Applied Biosystems, Foster City, USA). Protocols and primer sequences are available on request. Sequencing was performed on an ABI 3130xl genetic analyzer (Applied Biosystems).
Analysis of the MTHFR 677C>T variant (rs1801133): The MTHFR 677C>T genotype was determined by mutagenically separated PCR [38] using the protocol by Endler et al. [39] with slight modifications. PCR protocol and conditions are available on request.
Analysis of X-chromosome inactivation: The XCI pattern was determined by PCR analysis of the polymorphic CAG-repeat region in the HUMARA gene located next to a HpaII restriction site. Aliquots of undigested and HpaII-digested DNA were subjected to PCR using primer sequences and the protocol published by Mitterbauer et al. [40]. Amplicons were analyzed on an ABI 3130xl genetic analyzer using the Gene Scan 3.7 (ABI) software. For quantification, the areas under the peaks of the two alleles from the undigested and digested DNA were determined. In every subject the ratio between the peak area of both alleles was calculated, and the degree of X-chromosome inactivation is presented as ratio of the higher peak (X1) to the smaller peak (X2).
Statistical analysis
All statistical analyses were done using the PASW Statistics 18.0 software (SPSS Inc., Stanford, USA). Correlations between two continuous variables were calculated using the Pearson correlation coefficient (variables with a skewed distribution were transformed with the natural logarithm ln). An impact of XCI on FVIII levels was tested by a linear regression model. The association of the MTHFR genotype with the XCI pattern, FVIII:C and FVIII:Ag were evaluated by variance analysis including blood group and VWF:Ag levels as covariates. A pvalue <0.05 was considered statistically significant.
Demographic data, laboratory results and distribution of blood groups in carriers and controls are shown in Table 1.
| Carriers (n=61) | Controls (n=174) | |
| Age [years], median (range) | 45 (12-90) | 55 (20-84) |
| FVIII:C [%], median (range) | 75 (14-179) | 144 (71-237) |
| VWF:Ag [%], median (range)† | 116 (63-236) | 110 (55-236) |
| Blood group n (%)* | ||
| 0 | 18 (30.5) | 51 (34.7) |
| A | 25 (42.4) | 63 (42.9) |
| B | 10 (16.9) | 20 (13.6) |
| AB | 6 (10.2) | 13 (8.8) |
| Mutation type n (%): | ||
| Inversion intron 22 | 25 (41.0) | - |
| Inversion intron 1 | 1 (1.6) | - |
| Missense mutation | 18 (29.5) | - |
| Nonsense mutation | 6 (9.8) | - |
| Small deletion | 4 (6.6) | - |
| Splice site mutation | 7 (11.5) | - |
Table 1: Demographic data of hemophilia A carriers and controls. †VWF:Ag could be tested in 147 controls and all carriers; *blood group was not available in 2 carriers and 27 controls
X-chromosome inactivation and FVIII activity
The HUMARA assay allowed the distinction of the two Xchromosomes (X1, X2) in 54/61 (88.5%) carriers and 151/174 (86.8%) controls. For all these females the PCR products generated following HpaII digestion could be quantified by analysis on the ABI 3130xl, and the ratio of the two X-chromosomes could be determined. For statistical calculations the X1:X2 ratios were transformed to their natural logarithm (ln). We found that skewed imprinting occurs in healthy individuals and hemophilia A carriers. While the percentage of individuals with skewed imprinting was not statistically significantly different in carriers and controls (p=0.213), only 5/154 controls (3.3%) but 8/54 (14.8%) carriers had skewed XCI ratios of >1:2 (Figure 1). Interestingly, age was not associated with skewing (carriers: r=0.011, p=0.936; controls: r=0.066, p=0.423).
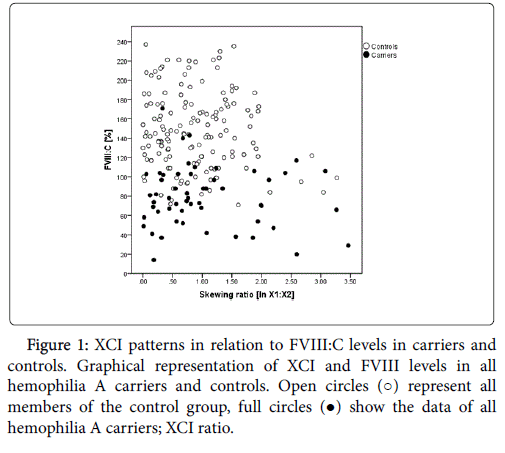
Figure 1: XCI patterns in relation to FVIII:C levels in carriers and controls. Graphical representation of XCI and FVIII levels in all hemophilia A carriers and controls. Open circles (○) represent all members of the control group, full circles (●) show the data of all hemophilia A carriers; XCI ratio.
In univariate analysis XCI was not correlated with FVIII:C in carriers (p=0.459, Figure 1) or controls (p=0.172, Figure 1). If VWF:Ag and blood group were included as covariates, skewed XCI was significantly associated with FVIII:C in controls (p=0.010) but not in carriers (p=0.365).
The comparison of individuals with an XCI ratio <1:0.33 (1:1.39 untransformed, n=13) with females with an XCI of 1:0.33 or higher (≥1:1.39 untransformed, n=41) showed median FVIII:C of 69% in individuals with an XCI<1:0.33 in contrast to a median FVIII:C of 78% in carriers of an XCI>1:0.33. Even though the differences were not significant (FVIII:C p=0.119) a trend for an influence of XCI on FVIII activity was visible.
In 26/54 carriers we could identify which of the two Xchromosomes carried the mutated F8 gene by family analysis, and we could determine the XCI status of this X-chromosome. In 13 females (group A) the intact F8 gene was located on the inactivated Xchromosome, in the other 13 females (group B) the intact F8 gene was located on the active X-chromosome. In group A the median XCI ratio was 1:2.67 (ln 1:0.98), in group B a median XCI of 1:2.40 (ln 1:0.88) was found. In group A the median FVIII:C was 73%, while group B had median FVIII:C of 88% (p=0.052). After inclusion of VWF:Ag and blood group (0 or Non-0) as covariates, a significant difference in FVIII:C (p=0.045) was observed.
XCI pattern and MTHFR 677C>T genotype
We found no significant association between the MTHFR 677C>T genotype and XCI (Tables 2 and 3) in carriers and controls.
| Controls (n=174)* | Carriers (n=61)* | |||||||
| MTHFR | n (%) | FVIII:C | p† | p‡ | n (%) | FVIII:C | p† | p‡ |
| CC | 79 (45.4) | 147 | 19 (31.1) | 97 | ||||
| CT | 72 (41.4) | 141 | 0.375 | 0.561 | 34 (55.7) | 73 | 0.033 | 0.031 |
| TT | 23 (13.2) | 136 | 8 (13.1) | 72 | ||||
| MTHFR | X1:X2 ratio | X1:X2 ratio | ||||||
| CC | 70 (46.4) | 01:00.9 | 17 (31.5) | 01:00.7 | ||||
| CT | 62 (41.1) | 01:00.7 | 0.673 | 0.558 | 30 (55.5) | 01:00.8 | 0.444 | 0.34 |
| TT | 19 (12.6) | 01:00.7 | 7 (13.0) | 01:00.7 | ||||
Table 2: Association of MTHFR 677C>T genotype with FVIII:C and XCI in carriers and controls. The numbers represent median values; XCI ratio was ln-transformed for statistical analysis; *151/174 controls and 54/61 carriers were heterozygous for the HUMARA locus; †pvalue calculated with variance analysis; ‡blood group and VWF:Ag as covariate.
| Inversion 22 Carriers (n=25) | Non-Inversion 22 Carriers (n=36) | |||||||
| MTHFR | n (%) | FVIII:C | p† | p‡ | n (%) | FVIII:C | p† | p‡ |
| CC | 7 (28.0) | 104 | 12 (33.3) | 79 | ||||
| CT | 13 (52.0) | 67 | 0.011 | 0.021 | 21 (58.3) | 81 | 0.391 | 0.044 |
| TT | 5 (20.0) | 65 | 3 (8.4) | 80 | ||||
| MTHFR | X1:X2 ratio | X1:X2 ratio | ||||||
| CC | 5 (23.8) | 01:00.7 | 12 (36.4) | 01:00.8 | ||||
| CT | 11 (52.4) | 01:00.8 | 0.897 | 0.735 | 19 (57.6) | 01:00.8 | 0.481 | 0.425 |
| TT | 5 (23.8) | 01:00.7 | 2 (6.0) | 01:00.6 | ||||
Table 3: Association of MTHFR 677C>T genotype with FVIII:C and XCI in Inv 22 vs. Non-Inv22 carriers. FVIII:C and XCI data are presented as median values; XCI ratio was ln-transformed for statistical analysis; †p-value calculated with variance analysis; ‡blood group and VWF:Ag as covariate.
The MTHFR genotype frequencies were distributed according to the Hardy-Weinberg equilibrium in both groups.
In controls with MTHFR 677CC median X1:X2 ratios of 1:2.38 (ln 1:0.87) were found while women with MTHFR 677TT had median X1:X2 ratios of 1:2.03 (ln 1:0.71; Figure 2a). In hemophilia A carriers with MTHFR 677CC or 677CT median X1:X2 ratios were 1:2.10 (ln 1:0.74), while carriers with MTHFR 677TT showed median X1:X2 ratios of 1:1.93 (ln 1:0.66; Figure 2a).
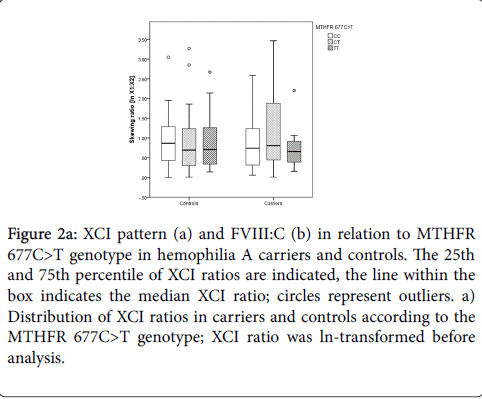
Figure 2a: XCI pattern (a) and FVIII:C (b) in relation to MTHFR 677C>T genotype in hemophilia A carriers and controls. The 25th and 75th percentile of XCI ratios are indicated, the line within the box indicates the median XCI ratio; circles represent outliers. a) Distribution of XCI ratios in carriers and controls according to the MTHFR 677C>T genotype; XCI ratio was ln-transformed before analysis.
MTHFR 677C>T genotype and FVIII activity
Hemophilia A carriers with MTHFR 677CC had median FVIII:C levels of 97%, while carriers with MTHFR 677TT showed median FVIII:C levels of 72% (Figure 2b).
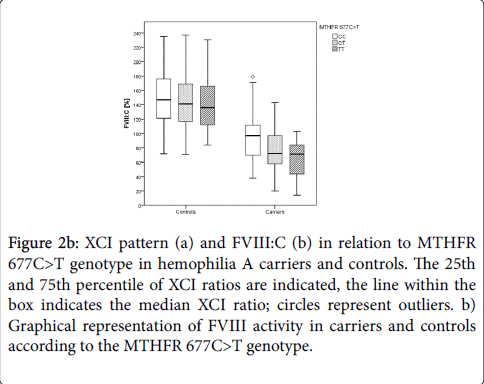
Figure 2b: XCI pattern (a) and FVIII:C (b) in relation to MTHFR 677C>T genotype in hemophilia A carriers and controls. The 25th and 75th percentile of XCI ratios are indicated, the line within the box indicates the median XCI ratio; circles represent outliers. b) Graphical representation of FVIII activity in carriers and controls according to the MTHFR 677C>T genotype.
The difference was statistically significant (p=0.033) and remained significant after inclusion of VWF:Ag and blood group as covariates (p=0.031, Table 2).
The association of the MTHFR 677C>T genotype and FVIII:C was particularly pronounced in carriers of an intron 22 inversion (p=0.011, Table 3). Controls carrying MTHFR 677CC showed median FVIII:C of 147%, while healthy females with MTHFR 677TT had median FVIII:C of 136% (Figure 2b). These differences in FVIII:C were not significant (p=0.375, Table 2).
FVIII activity is highly variable in carriers of hemophilia A and also in healthy individuals. von Willebrand factor levels, age and blood group are known modifiers of FVIII:C. In agreement with the literature, we found a significant correlation of VWF:Ag with FVIII activity in the control population and in carriers (unpublished data). However, blood group only had an effect on FVIII:C in healthy females (unpublished data). Age was not associated with FVIII activity neither in controls nor in hemophilia A carriers. Thus, we hypothesized that other factors function as additional modifiers of the FVIII activity in these women. It is known, that in some female hemophilia A patients highly skewed XCI patterns are associated with very low FVIII:C and with a hemophilia A phenotype [24-29]. Therefore, we suspected skewed XCI to be more frequently found in carriers than currently suspected.
Indeed, we observed a significant association of FVIII:C with XCI in healthy females when considering VWF and blood group as covariates. The association remained highly significant (p=0.008) even when only VWF was entered as covariate. This suggests that a skewed inactivation of the X-chromosome has an independent impact on FVIII:C in controls. In agreement with Orstavik and colleagues, we found no correlation between XCI and FVIII in confirmed hemophilia A carriers [41], even though 14.8% showed strong skewing of the Xchromosome. In controls, the number of females with strong skewing was much smaller and amounted to only 3.3%. It appears that the impact of a mutation in the F8 gene on FVIII:C is stronger than the effect of skewing. When we stratified the mutation carriers to two groups – those in whom the X-chromosome with the intact F8 was imprinted and those in whom the X-chromosome carrying the mutated F8 was imprinted we found lower FVIII:C in females in whom the X-chromosome with the intact F8 was imprinted. Correspondingly, when the X-chromosome with the mutated F8 was preferentially inactivated, FVIII:C levels were higher. The difference was borderline significant (p=0.052) which was probably due to the relatively small number of families that could be studied. When VWF:Ag was entered as covariate a significant association (p=0.015) was detected.
It is known that the MTHFR enzyme regulates DNA methylation by donating the essential methyl groups. The 677C>T polymorphism (rs1801133) reduces the activity of MTHFR. When we determined an association of the MTHFR genotype with XCI and FVIII levels, we found no significant influence of MTHFR 677C>T on XCI nor on FVIII levels in healthy females.
We observed a statistically significant association of MTHFR 677TT with lower FVIII:C in hemophilia A carriers. To our knowledge, this is the first study in which an association of the MTHFR 677C>T genotype with FVIII levels could be demonstrated in hemophilia A carriers. The MTHFR 677TT genotype was previously reported to slightly improve the clinical phenotype in severe hemophilia A patients [42-45]. In 2009 Ar et al. described a lower annual factor concentrate consumption in hemophilia A and B patients who carried a prothrombotic mutation - either factor V Leiden, factor II or a MTHFR genetic variant [46]. However, the difference was not statistically significant (p=0.203). Tüten et al. could identify a statistically significant difference (p=0.015) in factor utilization between patients with the MTHFR 677TT genotype compared to MTHFR 677 CC and CT carriers [47]. Moreover, hemophilia A patients with an inversion in intron 22 and with the prothrombin mutation PT20210A reported less severe bleeding episodes and a lower factor consumption per year [48].
Possibly, the results found in severe hemophilia A patients reflect an effect of the MTHFR genotype on VWF activity levels. Such an effect was shown by Tietjen et al. [49]. In his study, homozygous MTHFR 677TT individuals showed the highest VWF activity [49]. However, the association between the MTHFR 677C>T genotype and VWF levels is controversial. Perutelli et al. [50] did not observe a relationship of MTHFR 677C>T with VWF levels. In agreement with this study, we also could not find an association of the MTHFR genotype with VWF:Ag (p=0.706).
Interestingly, the association between MTHFR 677TT and FVIII activity was strong in carriers of an inversion in intron 22. It is known, that intron 22 of the F8 gene contains a 9.6 kb long CpG island. Methylation of these CpGs could differ depending on the MTHFR genotype. We attempted to prove this, however, the methylation status of the CpG island in intron 22 is very heterogeneous and not a suitable target to determine the imprinting of the F8 gene. This observation is in agreement with De Brasi et al, who came to the same conclusion [51].
The major limitation of our study is the small number of confirmed hemophilia A carriers that could be included. Nevertheless, we observed that the MTHFR 677C>T genotype was associated with FVIII:C levels particularly in females with an inversion in intron 22. We believe that our data are novel and are worthy to be confirmed in a multicenter study.
We have shown that XCI influences FVIII levels in healthy individuals after consideration of VWF and blood group. We have demonstrated that in female carriers of hemophilia A with a pronounced skewing and an inactivation of the X-chromosome with the intact F8, one has to expect low FVIII levels. Interestingly, we found clear evidence for an association of the MTHFR 677TT genotype with low FVIII:C levels in hemophilia A carriers, which was most pronounced in individuals with an inversion in intron 22.
We want to thank Renate Freitag, Department of Laboratory Diagnostics, Medical University of Vienna, for her help with mutation analysis of all hemophilia A carriers and Mag. Alexandra Kaider, Center for Medical Statistics, Informatics and Intelligent Systems, Section of Clinical Biometrics, Medical University Vienna, for her assistance with the statistical calculations. Further, the authors want to acknowledge the recruitment of carriers by Dr. Max Heistinger, Department of Internal Medicine I, Clinical Center of Klagenfurt am Woerthersee. The study was financially supported by the Austrian National Bank (project number 12208).