Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Research Article - (2014) Volume 2, Issue 6
We analysed between 1908 and 1985 of two hundred cases of thrombocythemia from the literature between 1908 and 1985. Erythromelalgic, acrocyanotic ischemia and digital gangrene, cerebral or coronary ischemic events (erythromelalgic thrombotic thrombocyhemia: ETT) in 99 thrombocythemia patients (essential thrombocythemia: ET N=67 and polycythemia vera: PV N=32) occured at platelet counts in excess of 400 × 109 /L. Hemorrhagic complications at time of presentation of hemorrhagic thrombocythemia (HT) in 100 HT patients included melana and/or hematemesis, skin bleedings (bruises, subcutaneous hematomas, painfull hematomas after trauma, ecchymoses, but not petechiae, epistaxis and gum bleeding and secondary bleeding after trauma or surgery. ETT occurred at an early stage of thrombocythemia with a mean platelet count of 1110+477 × 109 /L, whereas HT occurred at significantly higher platelet counts in excess of 1000 × 109 /L with a mean platelet count of 2016+1070 × 109 /L. HT patients frequently had a previous history of ETT or paradoxical occurrences of both thrombosis and bleeding (ETT/PHT). The degree of thrombocythosis determined the sequential occurrence of ETT, HT in ET and PV patients. Hypersensitive thrombocythemic platelets (sticky platelets) are involved in the etiology of platelet-mediated thrombosis at platelet counts above 400 × 109 /L whereas bleedings at high platelet count is related to an acquired von Willebrand syndrome processes on top of a blood clot retraction disturbance with erythrocyte fall out, that causes painful subcutaneous hematomas with a central swelling (clot) after a blow, trauma and secondary bleeding after surgery in thrombocythemia patients.
Keywords: Erythromelalgia; Hemorrhages; Arterial thrombosis; Thrombocythemia; Polycythemia vera; Essential thrombocythemia; Hemorrhagic thrombocythemia; Thrombotic thrombocythemia; Aspirin; Myeloproliferative disorders
Mitchel [1] reported on arare vaso-motor neurosis of the extremities and labeled it as erythromelalgia (erythros=red, melos=extremity, and algos=pain) in the footnote of his article. Mitchell demonstrated that typical erythromelalgia in one or more toes may progress into unbearable intense burning, throbbing, and aching pain, complicated by neurologic ischemic attacks and visual disturbances in one and the same patient. The occurrence of erythromelalgia in polycythemia is very well known in the literature since 1908 [2-5], (Norman and Allen). Osler in 1908 noted that there has been cases of PV in which pain in the hands and feet with the extreme congestion is suggestive for the erythromelalgia of Weir Mitchell [1]. In 1929, Oppenheimer recognized that the erythromelalgic symptom complex was often the earliest symptom of PV and even may precede PV for more than 10 years. He distinguished two types of peripheral circulatory symptoms in PV.1) the erythromelalgic syndrome, and 2) occlusion of peripheral arteries by thrombi (thromboangiitis obliterans) in later stages of the disease. Two of his patients suffered from erythromelalgia and recurrent attacks of dizziness and had platelet counts of 560 to 620 × 109/L. The peripheral microvascular symptoms at diagnosis of PV in 100 cases from the study of Brown and Griffin [4] were erythromelalgia in 6, acroparesthesias of burning type in 13, peripheral digital gangrene or ischemia in 8 (erythromelagic ischemic complications 21%). Similar observations of erythromelalgic microvascular ischemia, amourosis fugax, and transient ischemic attacks and major arterial and venous thrombotic events were observed in the studies of Edwards and Cooley [6], and of Barabas et al. [7] (Table 1).
| A. Vascular manifestations as the intial symptom of PV in 26 patientsaccording to Edwards abd Cooley 1970 | |
| Painful or unclean toes (good pulses in 6, fairly good pulses in 2 | 8 |
| Painful fingers andgood pulses | 2 |
| Major ischemia of a lower limb (with arteriosclerosis in 2) | 3 |
| Cerebral vascular disease (not specified) | 5 |
| Coronary heart disease (not specified) | 2 |
| Total | 20 |
| B. Vascular Complications in 200 PV patients according to Barabas et al. | |
| Cerebrovascular accidents | |
| Transient ischemic attack, facial weakness, or aphasia (TIA) | 26 |
| Permanent neurologic loss (Preceded by TIA I 6) | 13 |
| Attack of transient blindness (amaurosisfugax) | 4 |
| Peripheral artery disease | |
| Sudden ischemia of toe or finger(amputation of 1 or more digits in 10) | 15 |
| Femoral artery occlusion and amputated toe in the past | 3 |
| Femoral artery occlusion without preceding ischemia of the toes | 7 |
| Coronary heart disease (not specified) | 10 |
| Total arterial thrombotic complications | 78 |
| In 68 patients (34%) | |
| Superficial thrombophlebitis | 30 |
| Deep venous thrombosis | 26 |
| Splanchnic vein thrombosis | 5 |
| Pulmonary embolism | 4 |
| Priapism | 1 |
| Total venous thrombotic complications | 66 in 57 patients (28%) |
Table 1: Vascular manifestations.
In 1940 Dameshek and Henstell described recurrent burning pain and cyanosis of several toes of the right foot with gangrene of the third toe diagnosed as thrombo-angiitis oblitterans in PV at platelet count above 1000 × 109/L and erythrocyte count above 6 × 1012/L [8]. These observations prompted Dameshek in 1940 to suggest the possibility of “platelet thrombophilia” with multiple small peripheral vascular thromboses (thromboangiitis obliterans). In 1937 Nygaard and Brown [9] described a 47-year-old man with recurrent burning, painful, red toes during a 10-year follow-up ultimately complicated by blister formation and gangrene of the right fourth and fifth toes in a case with persistent increase of peripheral blood platelet count in myeloproliferative thrombocythemia. Annets and Tracy [10] and Vreeken and van Aken [11] described microvascular painfull ischemia of the toes as first manifestation of ET. Alarcon-Segovia et al. [12] recognized aspirinsensitive erythromelalgia as a clue to the early diagnosis of ET and PV, but phlebotomy did not improve the erythromelalgic burning pain (cases 5, 6, 7 in Table 2).
| Case | Age year | Platelets x109/L | Hb g% | Ht | Erythrocytes | Aspirine sensitive erythromelalgia | |
| x10/L | Diagnosis | ||||||
| 1 | 62 | 352 | 17 | 0.61 | 6.1 | PV | hands and feet since 6 years |
| 2 | 62 | - | 13 | 0.50 | 6.0 | PV | feet since 12 years |
| 3 | 68 | - | 19 | 0.74 | 8.1 | PV | feet since 2 years |
| 4 | 43 | 373 | 17.3 | 0.61 | PV | right foot since 5 years | |
| 5 | 70 | 1049 | 15.6 | - | 5.8 | ET/PV | feet since 14 years, phleb no effect |
| 6 | 62 | - | 14 | 0.51 | 5.1 | ET | right foot phlebotomy no effect |
| 7 | 61 | 837 | 17 | 0.62 | 6.3 | PV | left hand foot, phleb no effect |
| 8 | 57 | - | 12.4 | - | - | MPD | foot big toe developed MMM |
Table 2: Erythromelalgia as a clue to early diagnosis of myeloproliferative disorders (MPD), Alarcon-Segovia et al 1966.
Between 1970 and 1973 we discovered that aspirin responsive burning red extremities (erythromelalgia) is causally related to thrombocythemia in ET and PV patients (Figure 1). Acropatesthesias, for example tingling, “pins and needles” sensations, and numbness in the toes or fingers, usually precede the disabling and burning distress, and aspirin promptly relieves pain for approximately three days [13-15]. A few cases of erythromelalgia associated with primary (essential) thrombocythemia has been recognized in the 1970s [16- 20]. Between 1974 and 1977 Weatherley-Mein clearly documented that erythromelalgia was the presenting feature of primary (essential) thrombocythemia. Preston et al. [21] interpreted the burning painful and ischemic digital circulation disturbances as thrombotic thrombocythemia. Between 1975 and 1981 we demonstrated that aspirin responsive erythromelalgia is a pathognomonic manifestation of thrombocythemia in ET and PV patients is caused by plateletmediated inflammation and thrombosis in the end-arterial circulation (Figure 1). Aspirin relieves the peripheral, cerebral and ocular ischemic disturbances by irreversible inhibition of platelet cyclooxygenase (COX) activity and aggregation ex vivo and vitamin K antagonist (Warfarin), dipiridamol, ticlopedine, sulfinpyrazone and sofium salicytlate have no effect on platelet COX activity and are ineffective in the treatment thrombocythemia-specific manifestations of erythromelalgia and migraine-like ischemic attacks (MIAs) (Figure 2), [13-15]. Aspirin responsive erythromelalgia in thrombocythemia appeared to be utterly dissimilar from aspirin resisten incurable primary erythermalgia and secondary erythermalgia in SLE or RA patients [22-24]. Erythromelalgia disappeared by correction of platelet counts to normal (<350 × 109/L) in ET and PV, but not by correction of hemoglobin to normal by bloodletting in PV patients [13,15].
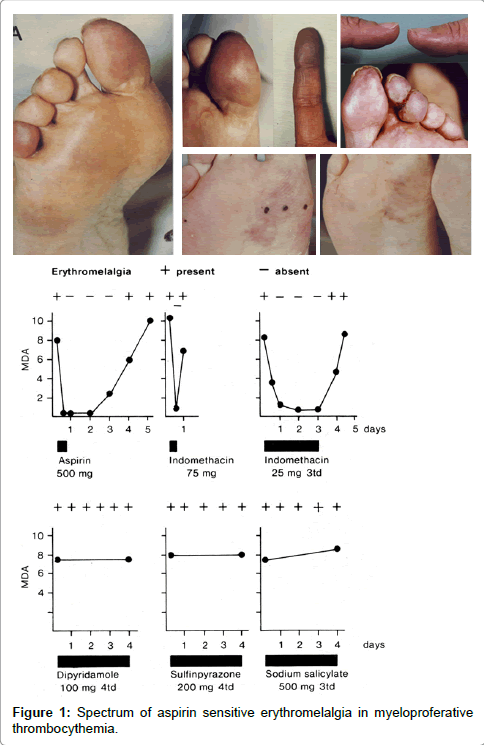
Figure 1: Spectrum of aspirin sensitive erythromelalgia in myeloproferative thrombocythemia.
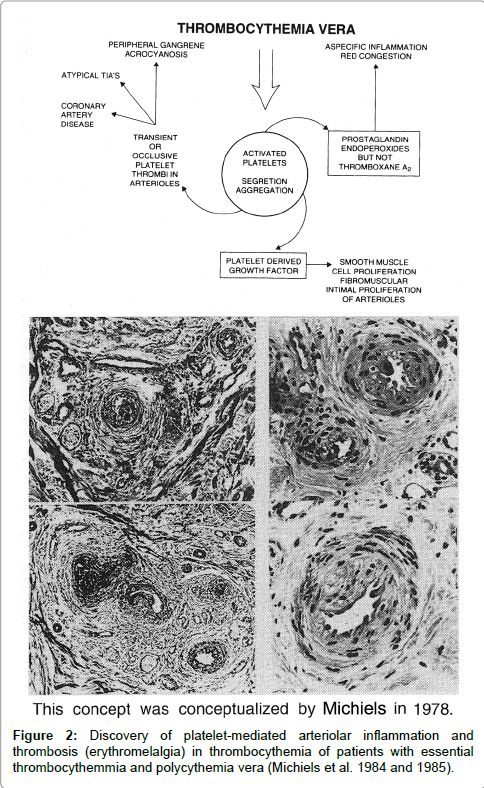
Figure 2: Discovery of platelet-mediated arteriolar inflammation and thrombosis (erythromelalgia) in thrombocythemia of patients with essential thrombocythemmia and polycythemia vera (Michiels et al. 1984 and 1985).
Minot and Buckmann recognized in 1923 [25] a bleeding tendency of ecchymosis, nose bleeds, and secondary bleedings but absence of pectechiae in association with persistent increased platelet count in a case of polycythemia vera. Rosenthal [26] recognized a group of post-splenectomy patients with persistent increase of platelet counts in excess of 1000 × 109/L associated with hemorrhagic or thrombotic complications. Thrombocythemia may be associated with both a hemorrhagic diathesis as well as thrombotic complications in one and the same patient [27,28]. Epstein and Goedel [29] introduced the term hemorrhagic thrombocythemia (HT) for a disease entity featured by recurrent gums and nose bleeds associated with very high platelet count of 1735 × 109/L and atrophic spleen in a 56 year old man This case was previously described as a case with “hochgradigen Thrombocytenvermehrung=high grade platelet increase” by Epstein and Kretz [30]. Similar case histories are described in the literature under a diversity of terms including hemorrhagic thrombocthemia (Uotila, Reid, Mortensen, Woodrow and Lope, Hardisty and Wolf, Binswanger et al., Koller and Bounameuax, Spaet and Bauer, Fountain , Gunz, shaw, Fountain and Losowsky, Webb et al., Cronberg et al., Hall, Ohler et al., Bensiger et al., and the Polycythemia Vera study Group, PVSG [31-45]) primary hemorrhagic thrombocythemia, [28,46-48] primary thrombocythemia [49], essential thrombocythemia [50-53], persistent thrombocythemia [54], thrombocythemia [55-57], postsplenectomy thrombocythemia [58], thrombocytosis [59], hemorrhagic thrombocytosis [60,61], myelose hyperthrombocytaire [62] and hyperplaquettose [63].
According to Reid [34], Fanger et al. [55], Ozer et al. [28] and Silverstein [48] HT may be part of or clearly associated with polycythemia vera, chronic myeloid leukemia, or myelofibrotic myeloid metaplasia. Others labeled HT as a distinct disease even when associated with other myeloproliferative diseases like polycythemia vera and myelofibrotic myeloid metaplasia [31]. According to Dameshek in 1950 [64], the cause of the hemorrhagic diathesis of polycythemia is obscure. Bleedings do not ordinarily occur spontaneously but only in response to trauma as with a blow or following operative procedures. Extreme degrees of postoperative hematomas are common, and excessive bleeding occurs after dental extractions, tonsillectomy, polypectomy and similar procedures. Based on the analysis of 50 cases from the literature and 5 of his own, Gunz [42] defined hemorrhagic thrombocythemia (HT) as clinical syndrome of recurrent spontaneous hemorrhages often preceded by thromboses, extremely high platelet count in excess of 1000 × 109/L, frequently splenomegaly, and hypochromic anemia with a tendency towards polycythemia between hemorrhages. Inclusion and exclusion
Bleeds from nose gums and gastrointestinal tract were most frequent followed by bruises and bleedings after trauma or surgery, whereas “thrombocytopenic purpura” were never seen. HT is obviously related to cases of primary or essential thrombocythemia, polycythemia vera, and agnogenic myeloid metaplasia (AMM). About half of the patients diagnosed as HT first come to medical attention via an emergency episode of acute bleeding or impending vascular occlusion. Spontaneous hemorrhages in HT typically develop at superficial cutaneous and mucosal surfaces, manifesting primarily as epistaxis, bruising, painful hematomas, ecchymoses, and bleeding from the gastrointestinal tract. Life threatening hemorrhage is infrequent and usually follows accidental or surgical trauma. When death occur, it was usually due to major thrombosis and much less frequent to hemorrhages. Deep tissue bleeding like joints bleeds seen in hemophilia, muscles hematomas, and intracerebral hemorrhage is very rare. Petechiae specific for thrombocythopathia of Glanzmann or Bernard Soulier or severe thrombocytopenia are never seen.
Vreeken and van Aken [11] described a patient with aspirinresponsive recurrent painful toes and fingers (erythromelalgia) in ET. Peripheral gangrene [21], microvascular ischemic or occlusive disease [18,65], and peripheral arterial thrombosis has been recognized as presenting symptoms of ET. Bernstein et al. [66] found ischemic signs on the electrocardiograms (ECG) in two ET patients with angina pectoris and “Prinz Metal” angina pectoris with transient increased of ST-segment on ECG) but a normal coronary angiogram has been observed in a 40-year-old man with ET [67]. Weber [68] reported a case of PV (hemoglobin 117%, erythrocytes 8.2 × 1012/L, and platelets 700 × 109/L) who had frequent attacks of ophthalmic migraine, which was associated with transien dilopia. Sometime there had been luminous visual phenomena, and at times he had transient attacks of giddiness. Neurologic manifestations in ET patients varied from recurrent attacks of transient ischemic attacks (TIA) to persistence of neurological deficits. Unilateral migraine-like headache, amaurosis fugax preceded by scotomas trigeminus neuralgia, aphasia, dysarthria, central facialis paresis, transient pareasis of one arm, leg or hemipareasis, persistent hemipararesis and organic vascular dementia have been frequently noted in ET patients [69-75]. Two ET patient became blind caused by thrombotic occlusion of the arteria centralis retinae. Singer [73] and Mundall et al. [75] observed attack of amaurosis vasospasm followed by white-yellow particle flowing from central to peripheral retina on fundoscopy, which disappeared together with complete recovery of the visus
Smith and Allen first discovered that a single dose of acetyl salicylic acid (aspirin) produced marked relief of the burning erythromelalgic distress that persited for a few days. Bab et al. [76] distinguished primary from secondary erythromelalgia associated with PV, systemic lupus erythromatodes (SLE) or rheumatiosd arthritis (RA) and stated that the response to aspirin is characteristic enough to constitute a valuable diagnostic clue for the diagnosis of erythromelalgia. Alarcon-Segovia et al. documented that aspirin-responsive erythromelalgia was a clue to early diagnosis in 8 PV patients associated thrombocythemia in 4 (Table 2), The spectrum of microvascular manifestations, cerebrovascular accidents, acute coronary artery syndromes superficial thrombophlebitis deep vein thrombosis and splanchnic vein thrombosis at time of diagnosis in the 1970s of 226 PV patients are shown in Table 3. Postsplenectomy thrombocytosis in MPD patients is typically persistent and complicated by bleeding and thrombotic complications [77-79]. Reactive thombocytosis is not associated with thrombotic or bleeding complications (McClure et al., Zucker and Mielke, Ginsburg, Coons et al.) [80]. The incidence of venous thrombosis is low in transient reactive post-splenectomy thrombocytosis [78,81,82]. Bleeding and thrombosis are rare in Ph+CML associated with persistent thrombocytosis [83].
| Diagnosis | ET | PV | ETT |
| Number of patients | |||
| Erythromelalgia without peripheral gangrene | 24 | 15 | 49 |
| Erythromelalgiaà peripheral digital gangrene | 23 | 8 | 31 |
| Peripheral gangrene (thromboangiitisobliterans) | 6 | 4 | 10 |
| Erythromelalagia and/or digital gangrene | 53 | 27 | 80 |
| Treatment for digital ischemia | |||
| Sympathecthomy | 4 | 1 | 5 |
| Amputation toe | 6 | 2 | 8 |
| Sympathectomy and toe amputation | 5 | - | 5 |
| Sympathectomy and/or amputation toe total | 15 | 3 | 18 |
| Angina pectoris | 6 | - | 6 |
| Myocardial infacrtion | 3 | 3 | 6 |
| Angina pectoris and/or myocardial infarction | 9 | 3 | 12 |
| Transient cerebral ischemic attack TIA | 3 | 3 | 6 |
| Amoaurosisfugax | 1 | 1 | |
| TIA àhemiparese | 2 | 2 | 4 |
| Hemiparese | 6 | 2 | 8 |
| TIA and/or hemiparese | 12 | 7 | 19 |
| Deep venous thrombosis | 3 | - | 3 |
| Pulmonary embolism | 2 | - | 3 |
| Superficial thrombophlebitis | 3 | - | 3 |
| Localisation of erythromelalgia and/or digital ischemia (thromboangiitisobliterans) in 80 cases with ETT (Michiels 1981) | |||
| Diagnosis | ET | PV | ETT |
| Toes | 35 | 17 | 51 |
| Toes/sole of the foot | 9 | 6 | 15 |
| Ankles | 3 | 3 | |
| Fingers | 3 | 1 | 4 |
| Toes and fingers | 4 | 4 | 8 |
| Total | 52 | 27 | 80 |
Table 3: Erythromelalgia and peripheral ischemic circulation disturbances in 99 cases with erythromelalgic thrombotic thrombocythemia (ETT) subdivided 67 cases with ET and 32 cases with PV from Tables 3 and 4.
Between 1975 and 1980, the author of this monography discovered that erythromelalgic thrombotic complications in thrombocythemia (ETT) occurred at slightly increased platelet counts (>400 × 109/L) in ET and PV patients, whereas hemorrhagic complications in thrombocythemia (HT) in myeloproliferative disorders tend to occur at platelet counts in excess of 1000 × 109/L. In order to document a relationship between platelet count and clinical complication of thrombosis or hemorrhages I reviewed in 1980 the spectrum erythromelalgic, thrombotic and hemorrhagic manifestations and hematological findings in 200 consecutive thrombocythemia cases published in the literature between 1929 and 1980. The 99 ETT cases histories from the literature could diagnosed as ET (N=67) and PV (N=32) and the 100 HT cases in thrombocythemia of various MPDs were reported in the literature between 1908 1980 and published in my thesis [13].
The platelet number in ETT patients subdivided in ET and PV andthe age distribution of 85 case histories of ETT is shown in Figure 3. Burning painful red or blue extremities, in retrospect very suggestive of erythromelalgia, were reported in 80 ETT cases. Peripheral (digital) gangrene developed in 31 cases. Eleven patients presented with peripheral gangrene without a record on burning. In 51 of the 62 evaluable cases, the peripheral arterial pulses were reported to be normal indicating the absence of significant atherosclerotic disease. Sympathectomy in 10 cases with digital gangrene (thromboangiitis obliterans) never induced improvement of the peripheral vascular insufficiency. Transient ischemic or thrombotic complications of the cerebral or coronary circulation were recorded in 12% and 19% of the cases, respectively. Deep vein thrombosis, pulmonary embolism and superficial thrombophlebiis of one or both leg rarely occurred (3% each).
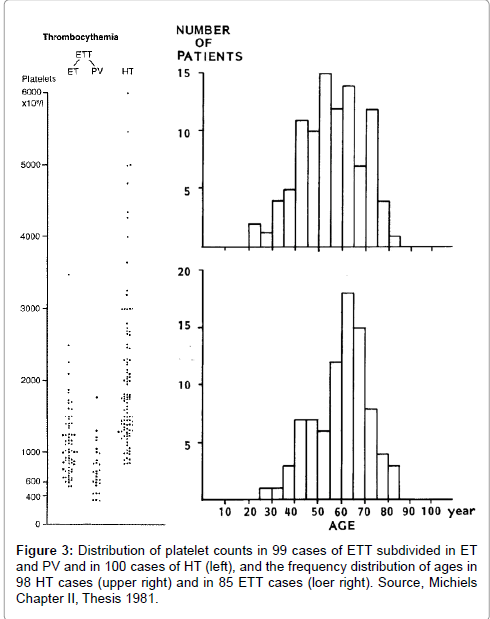
Figure 3: Distribution of platelet counts in 99 cases of ETT subdivided in ET and PV and in 100 cases of HT (left), and the frequency distribution of ages in 98 HT cases (upper right) and in 85 ETT cases (loer right). Source, Michiels Chapter II, Thesis 1981.
The hemorrhagic symptoms of 100 case histories from the literature between 1933 to 1978 are shown in Table 4. The age distribution of 98 HT patients is shown in Figure 3. Gastrointestinal blood loss usually occurred as melena and/or hematemesis. A bleeding source could not be detected, neither roentgenologically in 19 out of 24, nor gastroscopically in 8 out of 9 patients investigated. In a few cases, the bleeding source could not be localize even at laparotomy. Nose bleeds in 33 patients were generally profuse and relapsed frequently. The typical gingival bleeding in 15 patients usually occurred after tooth extraction. Skin bleedings were either recorded as bruises in 18 as subcutaneous hematomas in 15, and as ecchymoses or sugillations in 10 out of 34 patients. In some cases painful subcutaneous hematomas started with or had a central nodule (very likely a thrombotic clot), which extended to large hemorrhagic effusions is. Petechiae characteristic for severe thrombocytopenia or congenital thrombasthenia of Glanzmann were never seen.
| Cutaneous Bleedings | 34 PHT patients | |
| Bruises, bruising | 18 | |
| Subcutaneous hematomas | 10 | |
| Ecchymoses | 15 | |
| Nose bleeds | 33 | 33 HT patients |
| Gum bleeds | 15 | 15 HT patients |
| Secondary bleeds, trauma surgery | 29 | 29 HT patients |
| Gastrointestinal (GI) blood loss | 50 | 50 HT patients |
| Melena and hematemesis | 12 | |
| Melena | 17 | |
| Hematemesis | 6 | |
| Occult blood loss | 4 | |
| No details of GI blood loss | 11 | |
| Total type of bleeds in 100 PHT | 161 | 1.6 main type of bleeds per PHT patient |
Table 4: Main Type of Bleeding Manifestations in 100 reports of hemorrhagic thrombocythemia (HT).
Arterial and/or venous thrombosis preceded or followed the hemorrhagic periods in 15 out of 100 HT cases (15%) (Table 5). One HT case developed recurrent gangrene of the toes 4 years following the hemorrhagic manifestations [29,30]. The thrombotic manifestations preceding the hemorrhagic diathesis were described as painfull acrocyanosis in 3, gangrene of toes in 5, transient neurologic ischemic attacks in 3 and hemiparesis in 2, superficial thrombophlebitis in 3, priapism in 3 and deep vein leg thrombosis in one case only (Table 5). In 7 of 99 ETT cases (7%) hemorrhagic complications are recorded; subcutansous hematomas in 2, ecchymosis in 1, melena in 1 and secondary bleeding after surgery in 3 cases (Table 5).
| A.Preceding arterial or venous thrombosis in 15 out of 100 case histories (15%) of hemorrhagic thrombocythemia (HT) | ||||||||
| HT case | Author | Year | Type of thrombotic complication | |||||
| 1 | Epstein | 1933 | Recurrent gangrene of toes followed by toe amputation | |||||
| 2 | Uotila | 1938 | Transient hemiparesis, superficial thrombophlebitis left leg | |||||
| 3 | Mortensen | 1948 | Painful cyanosis of first and fifth right toes | |||||
| 4 | McGabe | 1955 | Painful cyanosis of fingers of the right hand | |||||
| 5 | Shaw | 1955 | Hemiparesis, peripheral circulation disturbances of one foot | |||||
| 6 | Hardisty | 1955 | Venous thrombosis left leg | |||||
| 7 | Hardisty | 1955 | Painful foot sole and ischemic ulcerations fifth right toe | |||||
| 8 | Hardisty | 1955 | Cerbral thrombosis, priapism | |||||
| 9 | Baikie | 1958 | Transient hemiparesis | |||||
| 10 | Gunz | 1960 | Superficial thrombphlbitis both legs | |||||
| 11 | Gunz | 1960 | Gangrene in fourth and fifth right toes | |||||
| 12 | Ozer | 1960 | Gangrene of the toes, priapism | |||||
| 13 | Ozer | 1960 | Burning pain in feet and finger, transient attacks of dysarthria | |||||
| 14 | Fountain | 1962 | Superficial thrombophlebitis of one leg | |||||
| 15 | Ougier | 1964 | Priapism | |||||
| B.The occurrence of bleeding complications in 7 of 99 reports of Erythromelalgic Thrombotic Thrombocythemia (ETT). | ||||||||
| EET case | Author | Year | Type of bleeding complication | |||||
| 1 | Moolten | 1949 | Subcutaneous hematoma | |||||
| 2 | Fanger | 1954 | Subcutaneous hematoma | |||||
| 3 | Shaw | 1961 | Ecchymoses | |||||
| 4 | Champion | 1963 | Melena | |||||
| 5 | Singh andWeitherley-Mein | 1977 | Secondary bleeding after surgery in 3 ETT cases | |||||
| 6 | Hussein | 1978 | Secondary bleeding after surgery in 3 ETT cases | |||||
Table 5: Bleeding Complications.
Post-splenectomy HT
The spleen in 64 evaluable ETT patients was not palpable in 27, slightly to moderately enlarged in 36 and greatly enlarged to below the costal margin in only 3. The spleen in 67 evaluable HT patients was not palpable in 17, slightly to moderately enlarged in 26 and greatly enlarged to below the umbilical level in 21 cases. In 2 ETT patients a splenectomy had been performed. In 20 HT patients, splenectomy was performed (spleen weighst in excess of 1 kilogram) had induced a post-splenectomy thrombocythemia complicated by bleedings (see section post-splenectomy HT) (Table 6). At time of splenectomy portal vein thrombosis was diagnosed in 2 and thrombosis of vena lienalis in 4 patients. Platelet counts in these 6 patients with splanchnic vein thrombosis was normal in 4 and strongly increased in 2. The majority of post-splenectomy HT patients (90%) had no bleeding manifestations at time of splenectomy. Platelet count at time of splenectomy was normal in 8, increased between 400-1000 × 109/L in 3, increased above 1000 × 109/L in 3, and not stated in 6 cases (Table 6). In the 3 cases with platelet counts of >1000 × 109/L had bleeding manifestation during or immediately after splenectomy (Table 6). In 17 post-splenectomy cases hemorrhagic manifestation became evident after 1 month in 4, after 3 months in 4, after 6 months in 1 and after one or a few years in 6 cases. At time of HT the platelet counts were far above 1000 × 109/L (range 1289-4353, Table 6). The weight of splenectomized spleen was significantly increased except in 1 and not stated in 4 (Table 6). One case of post-splenectomy HT in a 43-years old case was complicated by angina pectoris, myocardial infarction, and transient hemiparesis at platelet count of 1700 × 10/L [84], and by erythromelalgia (painful red swollen toes) at platelet count of 900 × 10/L in another case [85].
| Before Splectonomy | After Splectonomy | ||||||
| Author /Year | Bleeding symptoms | Platelets×109 | Bleeding symptoms | Months* | Platelets×109 | Spelen weight | |
| Brughs 1933 | - | 228 | + | 24 | 1735 | n.s | |
| Moolten 1949 | - | 580 | + | 3 | 2800 | 1100 | |
| Revol 1950 | - | 720 | + | 1 | 3655 | 600 | |
| Fanger 1954 | - | n.s | + | 36 | 4353 | n.s | |
| Hardisty 1955 | - | Normal | + | 1 | 1750 | >1000 | |
| - | n.s | + | 3 | 2735 | >1000 | ||
| Binswanger/Koller 1955/56 | - | 246 | + | 6 | 4050 | 800 | |
| Gunz 1960 | - | normal | + | 22 | 4000 | >1000 | |
| Ozer 1960 | - | n.s | + | 0 | n.s | 1000 | |
| Pederson 1961 | - | n.s | + | n.s | 2150 | n.s | |
| Fleisher 1962 | - | normal | + | 38 | 1289 | n.s | |
| Fountain 1962 | - | 145 | + | n.s | 1310 | 568 | |
| Ougier 1964 | - | 1470 | + | 0 | 1395 | >1000 | |
| Cronberg 1965 | + | 1920 | + | 0 | 1433 | 2200 | |
| Silverstein 1968 | - | n.s | + | 26 | 1312 | 280 | |
| Bensinger 1970 | + | 1000 | + | 0 | 1750 | 1030 | |
| - | normal | + | 1 | 3000 | 850 | ||
| - | n.s | + | 3 | 2000 | >1000 | ||
| - | 510 | + | 12 | 2300 | 1000 | ||
| Dube 1971 | - | 270 | + | 3 | 1300 | >1000 | |
Table 6: Post-splenectomy hemorrhagic thrombocythemia (HT) in 20 patients from the literature.
Platelet and leukocyte counts, and bone marrow findings in ETT and HT patients
Platelet counts ranged from 735 to 5000 × 109/L in HT and from 352 to 3500 × 109/L in ETT (Figure 3). The frequency distribution of the platelet and leukocyte counts in ETT and HT is shown in Table 7. The platelet count of 1059+507 × 109/L (mean+SD) in 96 ETT patients was significantly lower (P<0.001, Mann-Whitney-U test) than the platelet count of 2050+1107 × 109/L in 100 PHT patients. The mean leukocyte count of 25+17 × 10/L in 86 PHT patients was significantly higher than 14+6 × 10/L in 65 ETT patients (P<0.001). This leukocytosis in thrombocythemia is due to increased mature granulocytes. Half of HT cases had leukocytosis between 20 and 90 × 109/L. In some cases of HT the white blood cell differential count was left-shifted indicative for more advanced MPD disease [19,31,40,54,59,61,63].
| Plateletsx109/L | PHT | ETT | Leukocytes x109/L | PHT | ETT |
| <500 | 0% | 4% | 10-Apr | 10% | 23% |
| 500-1000 | 7% | 47% | 15-Oct | 21% | 46% |
| 1000-1500 | 32% | 31% | 15-20 | 14% | 19% |
| 1500-2000 | 21% | 14% | 20-90 | 55% | 12% |
| 2000-6000 | 40% | 4% | - | - | - |
| Mean x109/L | 2050 | 1059 | Mean x109/L | 25 | 14 |
| SD | 1107 | 507 | SD | 17 | 6 |
| Man-Whitney test | p<0.001 | p<0.001 | |||
Table 7: Frequency distribution of platelet and leukocyte counts in 100 cases of hemorrhagic thrombocythremia (HT) and in 96 cases of erythromelalgic thrombotic thrombocythemia (ETT). Source Michiels 1981.
The individual platelet in ETT and HT patients is shown in Figure 3. About half of 67 ETT patients had thrombotic complications at platelet counts between 400-1000 × 109/l (Table 8). The majority of symptomatic PV patients had similar microvascular thrombotic complications (ETT) at platelet counts between 375 and 1000 × 109/L (Figure 2). ETT usually persisted when the PV was brought into remission by phlebotomy simple because thrombocythemia persisted. There was no correlation between increased number of platelets and leukocytes in each of the categories of HT (r=0.07) and ETT (r=0.24).
| A.The 1980 RCP major (A) and confirmative (B) criteria for ET A1 Persistent platelet count in excess of 400x109/L. A2 Increase and clustering of enlarged megakaryocytes in bone marrow biopsy. A3 No or slight increase of reticulin fibers (RF 0 or RF 1) B1 Presence of large platelets in a peripheral blood smear B2 Absence of any underlying disease for reactive thrombocytosis and normal ESR. B3 No splenomegaly (<12 cm) or slight splenomegaly on palpation or scan (<15 cm) B4 Increase of LAP-score and no signs of fever or inflammation Exclusion criterion Ph+ chromosome and any other cytogenetic abnormality in blood or bone marrow cells |
| B. The 1980 RCP major (A) and minor (B) criteria for PV A1 Increased erythrocyte count above 6x1012/L: Dameshek 1940[42]. A2 Raised red cell mass. Male >36 ml/kg, female >32 ml/kg: PVSG 1971-1975 A3 Increase in bone marrow biopsy of clustered, large pleomorphic megakaryocytes with hyperlobulated nuclei and increased cellularity due to increased megakaryopoiesis erythropoiesis or typically trilinear mega-erythro-granulopoiesis. typical PV bone marrow excludes erythrocytosis. B1 Thrombocythemia, persistant increase of platelet >400x109/L B2 Leukocytosis, leucocyte count >109/L and low erythrocyte sedimentation rate (ESR) B3 Raised leukocyte alkaline phosphatase (LAP) score >100, absence of fever or infection B4 Splenomegaly on palpation or on isotope/ultrasound scanning A1or A2 plus A3 and none of B establishes erythrocythemic PV A1 or A2 plus A3 plus one of B establishes PV and excludes erythrocytosis |
Table 8: The Rotterdam Clinical and Pathological (RCP) criteria for Essential Thrombocythemia (ET) and Polycythemia Vera (PV) 1975-1980.
Bone marrow findings, mainly aspirates, in 36 HT and 31 ETT patients showed major increase of mature megakaryocytes and platelet clumps, normoblastic erythropoiesis and normal myelopoiesis thereby excluding erythroleukemia, preleukemia and MDS. In addition peripheral blood and bone marrow findings were consistent with chronic myeloid leukemia in 5 PHT and agnogenic myeloid metaplasia in 4 PHT patients. Between 1960 and 1970, Silverstein observed 47 HT patients in whom the thrombocythemia was associated with chronic granulocytic leukemia, myeloid metaplasia, or polycythemia vera in eight patients each.
At platelet counts in excess of 800 × 109/L, serum potassium concentrations are always significantly elevated in the presence of normal renal function and normal plasma potassium levels [86-89]. This pseudohyperkaliemia in thrombocythemia of various myeloproliferative disorders is explained by release of potassium from the platelets during coagulation and clot retraction [23,90].
The PVSG criteria in 1975 for the diagnosis of hemorrhagic thrombocythemia were and crude and required a minimal platelet count in excess of 1000 × 109/L [91] thereby overlooking symptomatic ET patients who presented with ETT at platelet counts above 400 × 109/L (Michiels et al. [15]). Focusing on the causal relation between erythromelalgia and thrombocythemia in ET and PV patients, we were able to document since 1975 the very early stage of ET by the use of the Rotterdam Clinical and Pathological (RCP) criteria for ET and PV [92,93]. The 1980 RCP criteria of ET and PV were determined by careful prospective documentation of peripheral blood and bone marrow smears and bone marrow biopsy material [93]. Platelets in excess of 400 × 109/L, and an increase of clustered enlarged megakaryocytes in a bone marrow biopsy material was found to be diagnostic for ET and excluded reactive thrombocytosis. On top of the clinical PVSG criteria for PV [46] we introduced in 1980 bone marrow histopathology and erythrocyte count above 6 × 1012/L proposed by Dameshek in 1940 [94] as specific clues to the diagnosis of PV to clearly differentiate PV from all variant of primary and secondary erythrocytosis. The 1980 RCP modifications of the 1975 PVSG criteria for PV include 4 main changes [13]. First, the major criterion O2-saturation of >92% is replaced by absence of primary or secondary erythrocytosis by clinical and laboratory tests. Second; splenomegaly is replaced by bone marrow histology as a major criterion (A3). Third, the 1980 RCP diagnostic set used splenomegaly as a minor criterion . Fourth, we skipped raised B12 (>900 ng/L) or raised B12 binding capacity (>2200 ng/L) as completely irrelevant for the diagnosis of early and overt stage PV.
The follow-up of the Rotterdam MPD Working Group (1975-1995) on the efficacy and safety for the prevention and treatment of thrombotic complications in 68 ET patients seen in the Academic Hospital Rotterdam has been critically evaluated by Perry van Genderen et al. [95,96]. The therapeutic implications from the prospective Rotterdam ET studies are completely in line with the concept that microcirculatory thrombotic complications in thrombocythemia already occur at platelet counts in excess of 400 × 109/L, which are relieved by reduction of platelet counts to normal (<400 × 109/L) or by control of platelet function with low dose aspirin. At time of presentation 54 patients had ET related microvascular thrombotic complications at platelet counts between 575 and 1031, mean 750 × 109/L and treated with low-dose aspirin. In patients receiving platelet lowering agents either busulfan or hydroxyurea and no aspirin, thrombotic complications occurred frequently during follow-up at platelet counts above 400 × 109/L ( 624 ± 255 × 109/L), indicating that inadequate control of platelet number in thrombocythemia of ET and PV patients is associated with a persistent high incidence of microvascular disturbances including MIAs when not on aspirin at plateletcounts above 350 to 400 × 109/L. One low dose aspirin reduces the risk of microvasular ischemic or thrombotic complications from above 50% to less than 5% during 6.2 years of follow-up, and thereby prevented the progression from low thrombotic risk to high risk thrombotic in ET and PV (Van Genderen et al. [95,96]). Aspirin in ET at platelet counts above 1000 × 109/l in general daily practices is not safe enough in terms of bleedings elicited by aspirin indicating the need to add non-leukemogenic platelet lowering agents. Thrombocythemia with platelet counts in excess of 1000+250 × 109/L is frequently associated with the paradoxical occurrence of thrombosis and bleeding. Mucocutaneous bleedings spontaneously occur at platelet count in excess of 1000+250 × 109/L due to an acquired type II-like von Willebrand syndrome (AVWS, absence of high and intermediate von Willebrand factor (VWF) multimers) increasing in severity at increasing platelet counts to high levels above 1000 to 1500 × 109/L (Figure 4, Van Genderen et al. [95]. Reduction of platelet count by anagrelide, interferon (or hydroxyurea if anagrelide and interferon fail) to less than 1000 × 109/L will result in the persistence of microvascular thrombotic events and the disappearance of the bleeding symptoms and AVWS (Figure 4). Correction of the platelet counts to normal is associated with no recurrences of microvascular events and complete correction of the VWF-multimeric pattern and correction of all VWFparameters to complete normal values.
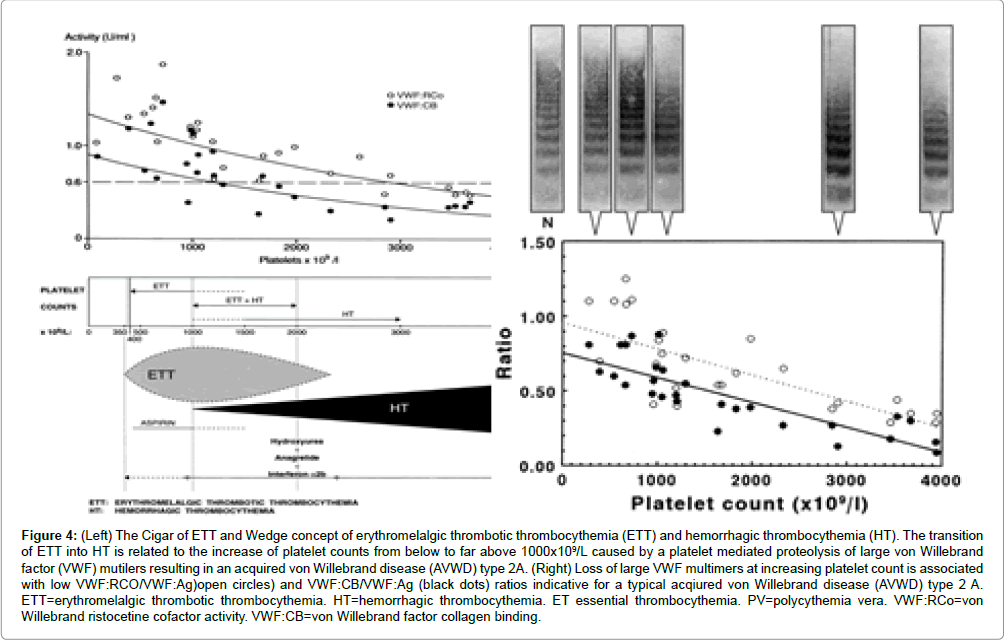
Figure 4: (Left) The Cigar of ETT and Wedge concept of erythromelalgic thrombotic thrombocythemia (ETT) and hemorrhagic thrombocythemia (HT). The transition of ETT into HT is related to the increase of platelet counts from below to far above 1000x109/L caused by a platelet mediated proteolysis of large von Willebrand factor (VWF) mutilers resulting in an acquired von Willebrand disease (AVWD) type 2A. (Right) Loss of large VWF multimers at increasing platelet count is associated with low VWF:RCO/VWF:Ag)open circles) and VWF:CB/VWF:Ag (black dots) ratios indicative for a typical acqiured von Willebrand disease (AVWD) type 2 A. ETT=erythromelalgic thrombotic thrombocythemia. HT=hemorrhagic thrombocythemia. ET essential thrombocythemia. PV=polycythemia vera. VWF:RCo=von Willebrand ristocetine cofactor activity. VWF:CB=von Willebrand factor collagen binding.