Journal of Clinical Trials
Open Access
ISSN: 2167-0870
ISSN: 2167-0870
Editorial Comment - (2013) Volume 3, Issue 5
1.1. Background: We have developed a new gene transfer vector based on nontransmissible recombinant Sendai virus expressing the human fibroblast growth factor-2 gene (DVC1-0101) to treat peripheral arterial disease. A phase I/IIa open-label four dose-escalation clinical trial for critical limb ischemia was completed. We concluded that DVC1-0101 is safe and well tolerated, and resulted in significant improvement of limb function. We present the protocol of the next phase of our study.
1.2. Methods: We plan to conduct a phase IIb clinical trial, which will be a randomized, placebo-controlled, parallel design, single-dose blinded and single center clinical trial in Japan. This study will enroll 60 patients diagnosed with PAD with intermittent claudication. Subjects who meet eligibility criteria will be randomized to receive a single dose of either placebo, 5 × 109 ciu/limb of DVC1-0101, or 1 × 109 ciu/limb of DVC1-0101 administered by direct intramuscular injection. The participation length in this trial for subjects will be approximately 12 months with nine visits. The primary endpoints are to evaluate the efficacy of DVC1-0101 versus placebo on peak walking time, and to evaluate the safety and tolerability of two dosage levels of DVC1-0101. The secondary endpoints are 1) to evaluate the effect of DVC1-0101 on claudication onset time, measured by a treadmill test and quality of life, measured using the Walking Impairment Questionnaire, 2) to determine the effect of DVC1-0101 on qualifying limb hemodynamics, and 3) to explore the pharmacodynamics of DVC1-0101 by evaluating biomarkers.
1.3. Discussion: The results of this trial will provide insights into the potential of DVC1-0101 for improving walking activities. The results will also help with the design of a possible phase III study.
Keywords: Sendai virus vector, Gene therapy, Fibroblast growth factor-2, Study protocol, Peripheral arterial disease, Intermittent claudication, DVC1-0101
In today’s aging society in Japan, the total number of deaths from atherosclerotic diseases (e.g., atherosclerosis obliterans, ischemic heart disease, and cerebro cardiovascular disease) exceeds the number of deaths from malignant neoplasms [1]. Therefore, there is an urgent need to take measures against atherosclerotic diseases to improve public health. The prevalence of Peripheral Arterial Disease (PAD), which is mainly due to atherosclerosis obliterans, is increasing in Japan. PAD is often asymptomatic or observed as numbness in the early stage (Grade I based on the Fontaine classification). However, as PAD progresses, patients’ Quality Of Life (QOL) decreases because of limited walking ability (Intermittent Claudication (IC), Grade II based on the Fontaine classification) associated with pain in the lower legs during walking. If untreated, IC progresses to Critical Limb Ischemia (CLI) in approximately one of four patients within 5 years [2], and induces pain at rest (Grade III based on the Fontaine classification) and ischemic ulcer/gangrene in the legs (Grade IV based on the Fontaine classification), resulting in substantial deterioration in the QOL. This affects the life prognosis of patients who are forced to be bedridden for a long time. This issue has become a cause of medical economic pressure associated with long-term medical care.
The prognosis of limb ischemia has greatly improved because of bypass surgery techniques, intra-postoperative management, artificial blood vessel material, endovascular treatment, and drug treatment. Based on the results of previous studies showing that the 5-year survival rate is approximately 40% for CLI and 70% to 80% for IC, the most important objectives for the treatment of PAD are (1) avoiding amputation and (2) preventing progression from IC to CLI [3]. However, in patients with chronic arterial occlusion who have bypass surgery to the popliteal artery or the lower leg, the 5-year patency rate is approximately 40%. This result suggests that such treatment is not yet sufficiently effective. In addition, there are many cases where bypass surgery is not indicated because of the presence of severe lesions in the peripheral region. Additionally, patients may not tolerate bypass surgery owing to a poor general condition associated with advanced arteriosclerosis in other organs. For CLI patients, mortality is as high as 20%, and major amputation is required within 1 year in 40% of patients [4]. There are many cases of complications with diabetes mellitus, which is an important risk factor of arteriosclerosis, because arteriosclerosis obliterans is a consequence of arteriosclerosis. Bypass surgery or endovascular treatment may not be indicated for patients with arteriosclerosis obliterans because they often have concurrent diabetic nephropathy requiring chronic dialysis. Additionally, they are often in poor health because of complications derived from diabetes mellitus accompanied by diffuse narrowing and advanced calcification. There is no effective treatment option available for such patients. Therefore, every year, tens of thousands of patients are estimated to gradually progress to CLI, resulting in amputation. The survival prognosis for patients who have had amputation is poor. According to the inter-society consensus for the management of PAD (TASC II), the mortality rate is twice as high as that for breast cancer, and is comparable with that of malignant tumors, such as colon cancer and Hodgkin’s lymphoma [3].
The most effective treatment of chronic arterial occlusion is surgery or endovascular treatment. However, there is no choice but drug treatment for patients for whom these invasive treatments are not indicated. Currently, there is no drug with proven efficacy for the treatment of CLI. Cilostazol and naftidrofuryl (not marketed in Japan) have been demonstrated to be effective in the treatment of IC in several large-scale clinical studies. Cilostazol is recommended as the first-line drug for the treatment of IC. Cilostazol has been reported to improve claudication distance by approximately 50%, but there is no evidence to support the effectiveness of cilostazol in preventing the progression from IC to CLI [3]. Given these circumstances, there is an urgent need to develop more effective techniques for the treatment of chronic arterial occlusion.
Therapeutic angiogenesis has been proposed [5] and clinically evaluated as a new treatment option for chronic arterial occlusion since the end of the 1990’s [6]. To date, techniques of therapeutic angiogenesis using (1) recombinant proteins, (2) bone-marrow/ blood cells, and (3) genes have been clinically tested. Sanofi-Aventis has been developing gene therapy products for the treatment of CLI using human Fibroblast Growth Factor-1 (FGF-1) expression plasmid. Additionally, Anges MG has been developing gene therapy products using Hepatocyte Growth Factor (HGF) expression plasmid. However, candidate drugs using recombinant proteins or genes have not yet succeeded in demonstrating efficacy in the treatment of IC.
In view of the current development of therapeutic angiogenesis techniques, we have attempted to investigate these problems through basic research. As a result, we have obtained the following findings from experiments using pathological animal models. (1) Gene expression is increased 50 to 500-fold by intramuscularly injecting the recombinant virus vector (SeV) that we developed compared with a technique using a plasmid. (2) The results of examinations comparing various genes related to angiogenesis as candidate therapeutic genes indicate that only the FGF-2 gene has a wide margin of safety without significant adverse drug reactions, and exerts a potent effect of relieving limb ischemia below the margin of safety. (3) FGF-2 activates the signaling system in different ways from other angiogenic factors. FGF-2 also strongly induces endogenous angiogenic factors, such as Vascular Endothelial Growth Factor (VEGF) and HGF downstream [7,8].
Based on the above data, researchers at Kyushu University Hospital planned a gene therapy clinical study in CLI patients with normal renal function using F gene-deleted non-transmissible recombinant Sendai virus vector expressing the human FGF-2 gene (rSeV/dF-hFGF2 [Development code: DVC1-0101]). Case registrations were started in April, 2006, after obtaining approval on January 31, 2006, from the Minister of Health, Labour and Welfare, and the Minister of the Environment in Japan (open-label, four-stage dose-escalation study corresponding to phase I/IIa in terms of study design). In total, 12 subjects, consisting of three subjects in the first stage (5 × 107 ciu/60 kg (ciu = cell infectious unit)), three subjects in the second stage (2 × 108 ciu/60 kg), three subjects in the third stage (1 × 109 ciu/60 kg), and three subjects in the fourth stage (5 × 109 ciu/ 60 kg), completed the scheduled administration of test product and observation period for 6 months. The gene therapy clinical study completion report was submitted to the Minister of Health, Labour and Welfare in Japan in March 10, 2011. The results suggested that DVC1-0101 can be used safely because it is well tolerated in patients and only affects the general condition of patients to a relatively small extent. At doses for the second stage (2 × 108 ciu/60 kg) or greater, DVC1-0101 appears to contribute to improvement of walking function and pain at rest among other efficacy endpoints [9].
The primary objective of the study is to investigate the safety and clinical efficacy of DVC1-0101 (1 × 109 ciu/leg, 5 × 109 ciu/leg) in patients with IC. We also aim to examine the dose-response relationship using the rate of improvement in walking function as an indicator.
Design
The DVC1-0101 trial is designed as a phase IIb investigator-initiated, randomized, centrally-registered, double-blinded, single center, and dose-response clinical trial of gene therapy versus placebo in patients with IC. The trial has a parallel-arm design with 1:1:1 allocation to the experimental intervention groups and the control intervention group. A full flow diagram for the study is shown in Figure 1.
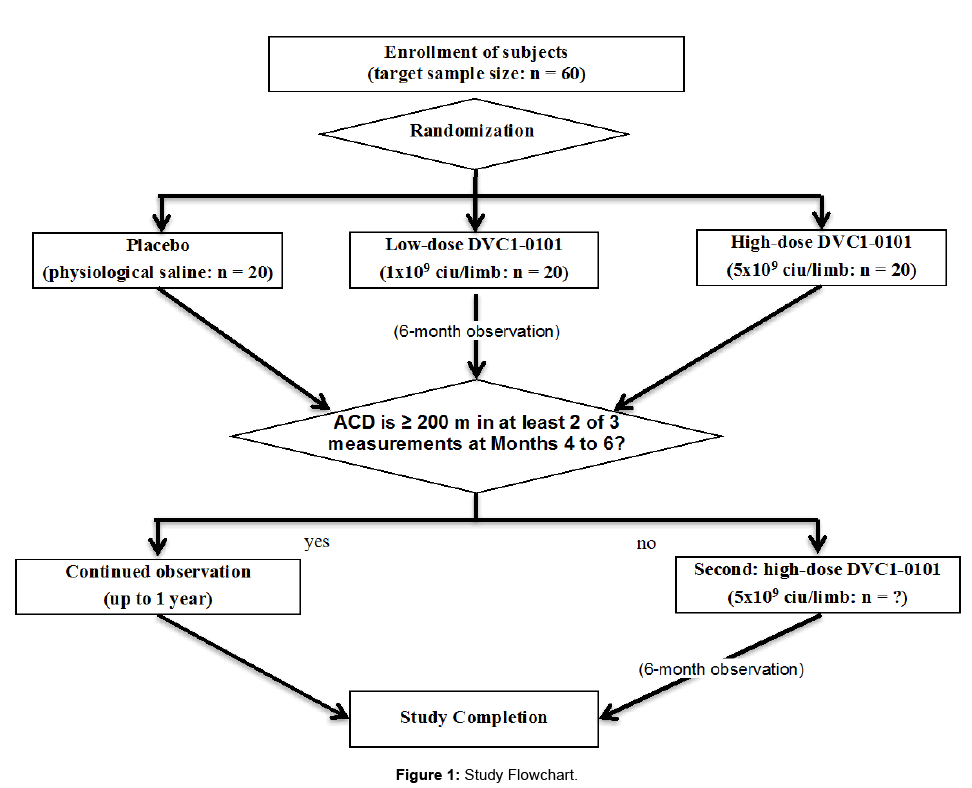
Figure 1: Study Flowchart.
Participants and setting
Eligible participants will include adults aged 40–80 years with arteriosclerosis obliterans accompanied by limited walking function due to IC (absolute claudication distance <200 m; Grade IIb and partly Grade III based on the Fontaine classification) [10]. The details of inclusion and exclusion criteria are shown in Table 1. A total of 60 subjects will have investigational product administered at a single center, Kyushu University Hospital, in Japan (Table 2).
| Inclusion Criteria | Exclusion Criteria |
|---|---|
| 1) Meet criteria (1) to (5) below and are confirmed as such by at least 1 specialist qualified by the Japanese Society for Cardiovascular Surgery and at least 1 specialist qualified by the Japanese Association of Cardiovascular Intervention and Therapeutics. (1) arteriosclerosis obliterans with stable symptoms, have intermittent claudication (ACD < 200 m) and are able to walk on a treadmill (2) resting ABI < 0.9 (3) refuse revascularization, risk of revascularization may be greater than the benefit, or develop obliteration after revascularization (4) angiographic findings show patency from the abdominal aorta through to the proximal side of the external iliac artery (5) angiographic findings meet the above criterion (4), and have stenosis or obliteration under the femoropopliteal region with morphology defined as type C or D based on TASCII 2) Administering cilostazol for at least 1 month and meet criterion 1). 3) Aged 40 years to 80 years. 4) Either sex, either inpatients or outpatients. 5) Able to give written consent for themselves. |
1) Have ischemic ulcer. 2) Diagnosed with Buerger’s disease. 3) Have a current or past history of life-threatening allergies. 4) Have been shown or are suspected to have cancer. 5) With concurrent proliferative intraocular neovascularization. 6) With concurrent cardiac failure (NYHA class II-IV). 7) With untreated severe arrhythmia. 8) Have or are suspected to have interstitial pneumonia. 9) Have progressive hepatic disorders. 10) Have moderate or severe hepatic disorders. (1) AST or ALT >2.5 times the upper limit (2) Prothrombin time is 14 seconds or longer (3) Serum bilirubin >2.0 times the upper limit 11) Diagnosed with hepatic cirrhosis (classified as B or C on the Child-Pugh). 12) Have an inflammatory disease. 13) Treated with immunosuppressants or corticosteroids for the treatment of various inflammatory diseases or after organ transplantation. 14) Underwent extirpative surgery of a malignant tumor in the past 5 years. 15) Have had a cerebral hemorrhage or cerebral infarction in the past 6 months. 16) With blood diseases. 17) With moderate or severe renal dysfunction CCr < 40 mL/min) 18) With alcohol or drug dependence. 19) Pregnant/lactating female, or who wish or are suspected to be pregnant. 20) Positive HIV antibodies. 21) Took part in any other clinical studies or research in the past 30 days. 22) Not permitted to participate in this study by the principal investigator or sub-investigator for any other reasons. |
Table 1: Inclusion and Exclusion Criteria.
| Procedures | Screening | Pre-admin | Admin | Day 1 | Day 2 | Day 3 | Day 5 | Day 7 | Day 10 to 14 | Month 1 | Month 2 | Month 3 | Month 4 | Month 5 | Month 6 | Year 1 | Year 2 | Year 3 | Year 4 | Year 5 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Informed Consent | X | |||||||||||||||||||
| Demographic and Medical History | X | |||||||||||||||||||
| Evaluation of Inclusion/Exclusion Criteria | X | X | ||||||||||||||||||
| Physical Exam | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | |
| Pre-treatment (antibiotics) | X | X | X | |||||||||||||||||
| Vital signs | X | X | X | X | X | X | X | X | X | X | X | X | ||||||||
| ABI/TBI | X | X | X | X | X | X | X | X | X | X | X | |||||||||
| Treadmill Familiarization | X | |||||||||||||||||||
| Exercise Treadmill | X | X | X | X | X | X | X | X | X | X | X | X | X | X | ||||||
| Angiography (IA-DSA) | X | X | ||||||||||||||||||
| Fontaine/Rutherford | X | X | X | X | X | X | X | X | X | X | X | X | X | X | ||||||
| VAS | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | |
| NIRs | X | X | X | X | X | X | X | X | X | X | X | X | X | X | ||||||
| WIQ(QOL) | X | X | X | X | ||||||||||||||||
| Eye Examination | X | X | ||||||||||||||||||
| ECG | X | X | X | X | X | X | ||||||||||||||
| Chest X-ray | X | X | ||||||||||||||||||
| Labs | X | X | X | X | X | X | X | X | X | X | X | X | X | |||||||
| Pregnancy Test | X | X | X | X | X | X | X | |||||||||||||
| Cancer Screening | X | X | X | X | X | X | X |
Table 2: Study Procedures.
Endpoints
In this study, a progressive load treadmill test shall be used to detect changes in walking function because this is more accurate than any other test [9]. The test condition is that the slope shall be progressively increased from 0 degrees by 2% every 2 minutes at a constant speed of 3.2 km/h. The Quinton® Q-STRESS® TM55 (Cardiac Science, Waukesha, WI, USA) will be used for the treadmill test.
Primary endpoints
• Rate of increase in absolute claudication distance (ACD)
• ACD
• Peak walking time
• Initial claudication distance (ICD)
• Claudication onset time
Secondary endpoints
• Measurement of oxygen dynamics in the leg muscles by nearinfrared spectroscopy after a treadmill load test
• Proportion of subjects in whom readministration was not required
• Evaluation of QOL based on the Walking Impairment Questionnaire (WIQ)
• Time-course changes using clinical stage classifications (Fontaine classification, Rutherford classification)
• Ankle-brachial pressure index
• Toe-brachial pressure index
• Time-course changes in pain at rest evaluated by the visual analogue scale
• Time-course changes in pain at rest evaluated by the frequency of analgesic use
• Incidence of cardiovascular events (to be followed up to 5 years after administration)
Procedures
Informed consent: The principal investigator or sub-investigator shall confirm that patients meet the inclusion criteria. They will directly provide patients with detailed information regarding participation in the study (including examinations to determine whether they meet the exclusion criteria) using the designated informed consent document for patients who wish to take part in the study. Subjects from whom consent is obtained shall be registered in the study and shall undergo screening tests.
Enrollment of subjects: The principal investigator or subinvestigator shall confirm that subjects from whom written consent to participate in this study is obtained meet the inclusion criteria and do not meet the exclusion criteria. They shall also record all the necessary information on the Enrollment Form and submit it by fax to the Enrollment Center. If registered subjects do not meet the inclusion criteria, or meet the exclusion criteria, the Enrollment Center shall inform the principal investigator or sub-investigator accordingly as soon as possible.
Randomization and blinding
The person responsible for investigational product assignment (assignment manager) shall inspect the investigational product for in distinguishes ability in appearance prior to assignment. The assignment shall be carried out using an assignment table prepared in advance by block randomization. The details of randomization (e.g., how to determine block length) shall not be disclosed.
This study shall use a double-blind design. Therefore, the result of investigational product assignment shall be concealed not only to subjects, but also to the principal investigator and sub-investigator. The control group (placebo group) will receive physiological saline (sodium chloride 0.9%) as a test product to maintain blinding. The study associate and laboratory staff shall not be informed of the details of treatment groups. In addition, data managers, statisticians, and all other persons concerned other than the assignment manager shall not be allowed to access the results of assignment. The assignment table shall be sealed and stored until unblinding. However, to ensure the safety of subjects, the results of assignment may be disclosed according to separately-specified emergency unblinding procedures.
The assignment manager shall disclose the results of assignment after confirming that data have been finalized for all subjects and blinding has been maintained during the study. Assignment shall be performed according to the separately-specified procedure manual, while reviewing the results of screening tests and checking the eligibility criteria after obtaining consent.
Intervention: The investigational product shall be prepared before use on the day of administration and used immediately after melting (within 8 hours). The vials filled with the investigational product will be placed in a double-sealed cooler and carried to the gene therapy unit approved to use the product according to the Type 1 Use Regulations of the Cartagena Law. This law is an act on the conservation and sustainable use of biological diversity through regulations on the use of living modified organisms (Act No. 97, 2003).
The principal investigator or sub-investigator shall observe which leg presents with symptoms faster during the treadmill load test to be performed 7 to 4 days before administration. The principal investigator or sub-investigator shall also determine the leg to be treated with the investigational product. On the day before administration, subjects shall be transferred to the gene therapy rooms approved to use the product according to the Type 1 Use Regulations of the Cartagena Law. The planned administration sites (30 sites in total: 15 sites in the upper leg and 15 sites in the lower leg) shall be marked with a permanent marker. Approximately 1 g of xylocaine 2% jelly will be applied on each site and be left for at least 15 minutes. The jelly will be removed and the leg disinfected with povidone-iodine. Anesthetic or analgesic will be used as required.
The investigational product will be drawn into a disposable 1 mL syringe using a 23G needle. A total of 0.5 mL of investigational product will be injected intramuscularly into each administration site (Figure 2). After administration, the administration sites will be wrapped with dressings.
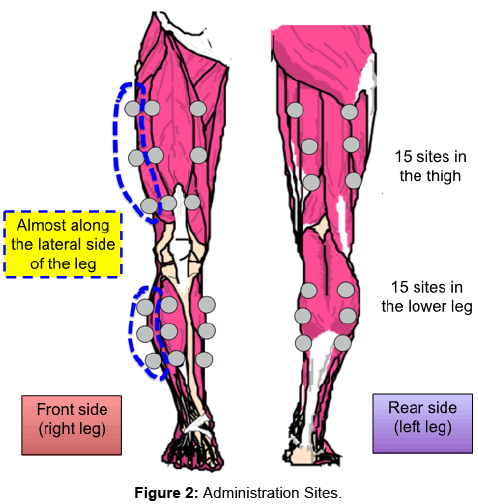
Figure 2: Administration Sites.
If the ACD is < 200 m measured using a treadmill load test at two or more visits in months 4, 5, and 6, the investigational product shall be given to subjects at a high dose (5 × 109 ciu/leg) for the second administration after obtaining consent again and confirming that they do not meet the exclusion criteria. Subjects who are shown to meet the exclusion criteria shall be reevaluated within 1 month from the visit in month 6 after the first administration. These subjects shall have the second administration at the time when they are shown not to meet the exclusion criteria.
Subjects shall be isolated and observed in the gene therapy rooms up to 7 days after administration. On day 7, the dressings will be removed and the subjects will be transferred to the general care unit.
Duration
The observation period is 6 months starting from the day of administration. Accordingly, the follow-up period of adverse events shall also be 6 months. However, if a serious adverse event occurs during the observation period and if it is not recovered from after the observation period, follow-up shall be performed at least bimonthly up to 5 years after administration.
Concomitant medication or treatment
Prohibited concomitant drugs: In principle, no prohibited concomitant drugs shall be specified. However, use of analgesics shall be prohibited in the 24 hours before the treadmill load test. Additionally, the use of platelet aggregation inhibitors (e.g., aspirin) and anticoagulants (e.g., warfarin) should be restricted during the week before and after administration, unless special circumstances arise.
Permitted concomitant drugs: No special restriction shall be applied. Concomitant use of vasodilators, peripheral circulationimproving drugs, platelet aggregation inhibitors, and other drugs that may affect the blood coagulation system shall be permitted for subjects who have used these drugs prior to the start of administration. However, the dosage shall not be changed unless special circumstances arise. After administration and during the observation period, the use of vasodilators, peripheral circulation-improving drugs, platelet aggregation inhibitors, and other drugs that may affect the blood coagulation system that had not been administered until that time shall not be permitted unless there is a particular reason for doing so.
Monitoring and quality assurance
The study monitor shall perform monitoring of the study institution on a regular basis according to the International Committee of Harmonization (ICH) guidelines for Good Clinical Research Practice (GCP). The study monitor shall confirm that the protocol and GCP are observed. The study monitor shall check the case report forms against the source documents to confirm that there is no inconsistency in the data recorded on the case report forms.
The person responsible for statistical analysis and person responsible for data management shall inspect the quality of data at each stage of data handling. This process is to guarantee the reliability and appropriate processing of data related to the study.
The person responsible for audit shall guarantee that the clinical study is conducted in compliance with GCP and the protocol by performing the audit. An on-site audit may be requested and performed by the researchers or designee personnel at any time.
Statistical analysis
Sample size determination: According to the results of the previous clinical study (corresponding to phase I/IIa study) preceding the planned present study, the log-transformed ratio of ACD before and after intervention was a mean of 0.5133 with a standard deviation of 0.3146 [9]. Because ACD improved by approximately 30% in the placebo group in the previous clinical studies, the log-transformed mean ratio of improvement in ACD (common logarithm) is estimated to be 0.1139. To demonstrate that improvement in ACD is more significant in the treatment groups than in the placebo group, a twosided t-test with a significance level of α = 5% will be used. At least 15 subjects will be required for each group to achieve a statistical power of 90%. Therefore, we intend to register a total of 60 subjects (20 subjects in the placebo group, 20 subjects in the low-dose group, and 20 subjects in the high-dose group) to allow for some ineligible subjects.
Primary outcome: The primary endpoint shall be the ratio of ACD before and after administration (rate of increase). Measurements shall be performed twice before administration and the higher value shall be used as the pre-dose value. Before administration, measurements shall be performed twice at the time of screening and during the period from 7 to 4 days before administration. The geometric mean of values measured at each visit during the period from 4 to 6 months after administration shall be used as the post-dose value. Missing data shall be handled on the basis of the last observation carried forward. The ratios before and after the study shall be calculated for each subject. This ratio shall be a maximum of 10.
For the primary endpoint, log-transformed values shall be compared using the least significant difference test. Among the placebo, low-dose, and high-dose groups, the mean difference in log-transformed values of the primary endpoint and the 95% confidence interval shall be estimated using least square means. The results shall be shown as geometric means. For values measured at 6 months after administration, the mean difference in ACD and its confidence interval shall be separately estimated based on several measurements for the group of subjects who switched to a high dose (re-administration group) and for the maintenance dose group. The details of analysis items and methods shall be specified in the statistical analysis plan to be prepared separately.
The primary analysis shall be an intention-to-treat analysis. A per protocol analysis shall also be performed as a secondary analysis.
Secondary outcomes: Secondary endpoints shall be analyzed for the purpose of discussion to support the analysis of the primary endpoint. Multiple comparisons shall not be performed because the analysis of secondary endpoints is exploratory. The details of the analysis method shall be specified in the statistical analysis plan to be prepared separately.
Safety analysis: The incidence of adverse events and abnormal clinical laboratory findings shall be compared between the groups. Adverse events and the severity of abnormal clinical laboratory values shall be defined according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. The details of the analysis methods shall be specified in the statistical analysis plan to be prepared separately.
Safety
From the time of obtaining written consent to the visit in month 6 after administration during the observation period, all adverse events that occur during this period shall be recorded on the case report forms. However, serious adverse events that require follow-up may have to be reported even after the observation period.
The principal investigator must evaluate the causality for all adverse events. Therefore, the principal investigator shall record the causal relationship between the adverse event and the investigational product on the case report form. Serious adverse events shall be evaluated according to the reporting requirements. The principal investigator shall determine whether the investigational product may have reasonably caused the adverse event or contributed to its occurrence. If the principal investigator determines that a serious adverse event is related to the study procedures, this investigator must record the causal relationship in the source document and case report form, and report the event if it meets the reporting requirements. In the case that a serious adverse event occurs, the principal investigator shall make contact with the hospital director within 24 hours after finding out about the event. The reporting procedures shall comply with the separately-specified Handling Manual for Safety Information. The principal investigator shall perform a follow-up for all serious adverse events, and submit a report detailing the obtained information to the hospital director before the above-mentioned deadline.
Ethical considerations
This study shall conform to the spirit of the Declaration of Helsinki to ensure the human rights, welfare, and safety of subjects to the maximum extent. The study shall respect the privacy of subjects and ensure confidentiality in preparing the case report forms, and in allowing direct access to and handling of the source documents. The trial will be conducted in compliance with the guidelines of gene therapy in Japanese law, the ICH-GCP, and the Cartagena Law. The trial will be started after receiving approval from the Institutional Review Board of Kyushu University.
The principal investigator must provide subjects with a thorough explanation of the contents and purpose of the study, and possible risks posed by the study. Prior to working on the operations of the study, the principal investigator must obtain written consent from each subject, and maintain the original written consent with the signature of each subject.
Publication policy
The copyright on works to be created and published based on the results of this study shall belong to Kyushu University Hospital. The information (unpublished data) contained in this protocol shall not be disclosed to any third parties without obtaining approval from Kyushu University Hospital.
The proprietary right on data to be obtained in this study shall belong to Kyushu University Hospital. Publication in part or whole of the study results externally at conferences or in journals (except for disclosure to regulatory authorities) shall require prior approval from Kyushu University Hospital. When publishing the study results, due consideration must be given to subjects’ privacy. Information must be published in a way that all personal identifiers are masked.
The conduct of this study shall be registered in the University Hospital Medical Information Network (UMIN), Clinical Trials Registry (CTR), or UMIN-CTR, and the information shall be disclosed.
The outcomes of this research will provide valuable proof of concept data for a future study of DVC1-0101 with PAD patients. Data from this study may be used to inform the design of a phase III study, and will lead to a better understanding of the gene therapy paradigm.
Comparable drugs
Disabling IC progresses to CLI in a relatively high proportion of cases. Surgery and endovascular treatment have been proven to be the most effective treatments in improving walking function. However, these treatments are not indicated for severe calcification and diffuse peripheral lesions in many cases. In Japan, cilostazol has been used as the first-line drug for CI, and there is accumulated evidence for its efficacy. However, cilostazol is effective in improving claudication in only approximately 50% of patients when using ACD as an indicator of therapeutic effect [11]. Currently, there is no medication that improves the walking function of patients by disabling IC to Grade IIb or below according to the Fontaine classification. Therefore, it is important to develop such medications to prevent progression to CLI in patients with IC.
Our study on DVC1-0101 will be the first to evaluate the efficacy of rSeV/dF-hFGF2 with PAD. DVC1-0101 is a recombinant preparation developed on the basis of the Sendai virus, which is classified in the group of murine parainfluenza viruses. DVC1-0101 has demonstrated significant therapeutic effects in nonclinical efficacy studies, and no toxicity has been observed within the therapeutic window. In the clinical study (corresponding to a phase I/IIa open-label study) conducted at Kyushu University Hospital in CLI patients, there were no adverse events reported evidently associated with the use of DVC1- 0101, but improvement in pain at rest and IC was observed in several patients [9]. In terms of improvement in ACD, DVC1-0101 showed much better results than cilostazol, as shown in a meta-analysis of cilostazol [11] (improvement of approximately 254% on average at 6 months after administration [except for Buerger’s disease]), and it is expected to be an effective treatment for CLI.
The results of a previous clinical study (corresponding to a phase I/IIa study) showed no serious adverse events with a direct causal relationship to the use of DVC1-0101 [9]. These results suggest there is no issue with safety in using DVC1-0101 at doses to be administered in the clinical study being planned. In addition, it has been demonstrated that single-dose administration of DVC1-0101 reproducibly improves pain at rest and ACD/ICD, among efficacy endpoints. In terms of the endpoint of ACD/ICD in particular, DVC1-0101 tends to provide much better results than cilostazol (Pletaal, Otsuka Pharmaceutical Co., Ltd.)
Safety issues
Expected adverse drug reactions in association with overexpression of angiogenic factors (human FGF-2) are a major concern. Overexpression of angiogenic factors, VEGF in particular, sometimes leads to hemangioma in the body (administration sites), as reported by several research groups, including Kyushu University [12-14]. Vascular abnormalities, such as hemangioma, have not been observed in association with FGF-2 overexpression by rSeV/dF-hFGF2 or in FGF-2 transgenic mice [15]. Therefore, these findings suggest that the risk of developing hemangioma in this study is low. In principle, a waitand- see approach shall be taken, even in the event of hemangioma, because it is a benign lesion. When the risk of rupture or bleeding is increased as hemangioma rapidly grows resection by percutaneous embolization or surgery is recommended.
FGF-2 protein expression, as well as other angiogenic factors, promotes the growth of malignant tumors. Therefore, even if efforts are made to rule out the presence of malignant tumors using detailed total body examinations (e.g., total body Computed Tomography (CT)), the presence of latent cancer may be undetected because of limited detection sensitivity of various examinations. However, studies in mice and monkeys found that FGF-2 expression was not detected in blood, even when human FGF-2 protein was overexpressed by rSeV/ dF-hFGF2 in skeletal muscle in the leg (detection sensitivity <5 pg/ mL) [15]. Particularly in the study using mice, FGF-2 protein was not detected in blood, even when human FGF-2 protein was overexpressed to a degree where the local intramuscular concentration reached 300 times that of endogenous FGF-2 protein. Therefore, FGF-2 protein is thought to bind to the extracellular matrix and remain in the vicinity of the administration site, based on biological characteristics of FGF-2 protein. Total body examinations, such as CT and tumor marker tests, shall be performed before starting the treatment.
For the same reason as mentioned above, the exacerbation of proliferative diabetic retinopathy cannot be excluded because it is a progressive disease. In the preceding clinical study, no findings were obtained that suggested the exacerbation of retinal disease accompanied by angiogenesis [9]. Patients with retinal diseases accompanied by angiogenesis, such as proliferative diabetic retinopathy, shall be excluded from the planned study. At the pretreatment evaluation stage, subjects shall be screened using funduscopy by an ophthalmologist.
Angiogenic factors may promote arteriosclerosis. For VEGF in particular, animal models have shown that even a low dose of VEGF in blood significantly promotes the progress of arteriosclerosis [16,17]. In humans, the severity of arteriosclerosis in the carotid artery correlates with blood VEGF concentrations [18]. Acid FGFs (aFGF/FGF-1), members of the FGF family, relate to the severity of arteriosclerosis, but there is no relation with FGF-2 [18]. However, the possibility cannot be excluded that arteriosclerosis progresses by indirect induction of HGF and VEGF in association with overexpression of FGF-2 [19]. In our preceding clinical study [9], no findings were obtained suggesting the accelerated progress of arteriosclerosis. In our planned study, subjects shall undergo follow-up examinations at outpatient visits on a regular basis after discharge for up to 6 months after administration. These examinations will include cardiovascular system examinations and total body CT (head, chest, and abdomen) in months 1 and 6 after administration to monitor the progress of arteriosclerosis.
Quality of measurements
The treadmill load test is internationally recommended as an established technique to evaluate efficacy endpoints in patients with disabling IC. However, some placebo effects are often observed in the evaluation of disabling IC. Therefore, there is a consensus among international specialists to recommend carrying out a parallel-group, randomized, double-blinded, placebo-controlled study to evaluate efficacy of the test product [20]. To overcome this issue, we shall use the Endpoint Quality Intervention Program (EQuIP), developed by CPC Clinical Research. This program will standardize data collection methods and assess results for integrity, data with low variability, and decreased placebo response, creating a decisive development path [21,22]. We shall use EQuIP in the treadmill test for ankle-brachial pressure index/toe-brachial pressure index measurements, and near-infrared spectroscopy. Trained personnel who have received a treadmill examiner certificate from CPC will perform an actual exam for the participants.
Trial status
The study design was agreed upon with the Pharmaceuticals and Medical Devices Agency, Japan in 2011. The protocol will be submitted to the Institutional Review Board at Kyushu University Hospital on September 2013. This study will be carried out in compliance with Types 1 and 2 Use Regulations of the Cartagena Law. Patient recruitment will start in March 2014. The study will finish enrolment as soon as 60 patients are included, and is expected to finish by March 2016. Collection and analysis of the results will be performed in the following 2 years.
The authors thank Yoko Nakamura for her help with the trial paperwork, and the Center for Clinical and Translational Research at Kyushu University Hospital for their management of this trial. This study will be supported in part by a Health Labour Sciences Research Grant to YY. Dr Yonemitsu is a member of the Scientific Advisory Board of DENAVEC Corporation. The other authors declare no conflict of interest.