Fungal Genomics & Biology
Open Access
ISSN: 2165-8056
ISSN: 2165-8056
Research Article - (2014) Volume 4, Issue 1
Keywords: Trichoderma asperellum, transcriptome, CAZymes, Peptide Pattern Recognition, plant cell wall degrading enzymes(CWDE)
In recent years a significant effort has been put into efficient utilization of plant biomass, such as of agricultural crops, crop residues and industrial byproducts, for conversion into a range of value-added bio-products to maximize the value derived from the biomass feedstock [1,2]. Plants have evolved complex structural and chemical mechanisms for resisting degradation of their structural sugars by for example fungi [3] To overcome the problem of recalcitrance of the plant cell wall, the industry uses a range of pretreatment approaches such as use of cell wall degrading enzymes to convert the plant cell wall polymers into smaller molecules [4]. In nature, various microorganisms produce enzymes that act independently or in synergy to break down the plant cell wall. Although it is not fully known how many enzymes are involved in plant cell-wall degradation, three general categories of enzymes are considered necessary to hydrolyze plant cell-wall materials: Cellulases, hemicellulases, and accessory enzymes. The recalcitrance of plant cell walls to enzymatic degradation and the high cost of the necessary hydrolytic enzymes are significant barriers to the global and large-scale production of biofuels and value-added bio-based products [5]. There is a need to develop more efficient and cost effective enzyme mixtures for the conversion of biomass to fermentable sugars to increase production of cellulose-derived value-added products, such as food, feed, chemicals and biofuels. To address this challenge, it is essential to gain a better understanding of the interactions between plant cell wall polysaccharides and cell wall degrading enzymes (CWDE). The complexity of the plant cell wall is mirrored by the diverse arsenal of CWDE produced by lignocellulose-degrading microbes. Enzymes acting on sugar structures are collectively called Carbohydrate-Active enzymes [CAZymes] [6]. The diversity of CAZymes reflects the structural diversity of plant cell walls. Each type of CAZyme function is represented in multiple families determined by sequence and structural similarities [7-9]. There are currently 132 glycoside hydrolase (GH) families in the CAZy database, http://www.cazy.org/. As a consequence of more genomes being sequenced a large number of enzymes are either labelled as “hypothetical” or even annotated incorrectly. High throughput annotation of CAZYmesis done largely based on GH family using Blast or HHMer programs, leaving many proteins without a functional annotation [10]. A more specific annotation is achieved by a following manual analysis as for example described for the reannotation of Trichoderma reesei CAZY genes [11].
Many Trichoderma species are strong opportunistic invaders. They are fast growing and abundantly producers of spores, and contain highly active CAZymesand secondary metabolites [12]. T. asperellum is a mycoparasitic species that are well known and widely used for their ability to inhibit the growth of plant pathogens. Their multi-enzymatic systems, composed of chitinases [13], ß-glucanases [14,15] laccases[16] swollenins [17] and proteases [18] position them as some of the best anti-microbial agents [19-21]. In addition to anti-microbial capability, T. asperellum has shown its potential in secreting other GHs under appropriate culture conditions [14,22] such as on cellulose [23] and on sugar bagasse [22]. On the latter substrate, T. asperellum showed promising results as an enzyme producer with a higher diversity of hemicelluloses and β-glucosidases compared to T. reesei.
The purpose of the present work was to analyze the GH enzymes in the transcriptome of T. asperellum when grown on wheat bran. This was done partly to understand the plant cell wall degrading system of T. asperellum and to identify enzymes important for the degradation of complex sugar structures, as well as to discover new enzymes. Several Trichoderma genomes are now available, which permitted a comparative study of GHs with the bioinformatic tool Peptide Pattern Recognition (PPR). With PPR it is possible to predict not only the GH family but also the enzyme function, so that the CAZymes may be compared on a functional level [24].
Strains
Trichoderma asperellum, strain number CBS 433.97 from the Centralbureau, Schimmelcultures, CBS, The Netherlands, was used in this study. For genome comparison the following genome sequences were used: Trichoderma reesei (Genbank accession numberGCA_000167675.2); Trichoderma atroviride (GCA_000171015.2); Trichoderma virens (GCA_000170995.2); Trichoderma hamatum (GCA_000331835.1);Trichoderma longibrachiatum (GCA_000332775.1)
Biomass
T. asperellum was cultivated on 1.5% (w/v) agar plates (Sigma Aldrich, UK) with 2.5% (w/v) different cellulosic material for 3, 5 and 7 days each at 200C and 300C, where additionally each combination of incubation duration and temperature was conducted at three levels of pH (pH 4, pH 6.5 and pH 8). Cellulosic material included: wheat bran (Finax, Denmark), oatmeal (Kornkammeret, Denmark), Spirodella polyrrhiza (BD-D2013-7, CIB, Chengdu, China), Lemna minor (BDL2013- 7, CIB, Chengdu, China), wheat straw, Brassica oleracea var, medullosa, Sinapis alba, Cannabis sativa [25].
Activity
Agar plate cultures of T. asperellum on each of the above cellulosic biomass substrates werewashed with 10ml H2O containing 1% Tween 80 to extract secreted enzymes. Enzyme activity was measured on AZCL-plates prepared using 1% agarose in buffer, pH 5.8, containing 0.1% azurine cross-linked (AZCL) substrates (Megazymes, Bray, Ireland) as described in the protocol. Inoculation was done by placing 15μL of enzyme blend from the growth plates in holes punched in AZCL-agarose plates. Enzyme activity was indicated by the area (cm2) of blue color zones resulting from hydrolysis of the substrate. The extent of the blue zones, indicating the enzyme activity, was measured after 24h at 300C.
RNA isolation
Mycelia were scraped from the surface of two agar plates on which T. asperellum had grown on wheat bran for seven days at 30°C, frozen in liquid nitrogen and ground to a powder. RNA was extracted from the mycelia with fenozol and the RNA Total Maxi kit protocol (A&A Biotechnology, Gdynia, Poland).
Transcriptome sequencing
RNA was sequenced by Beijing Genomics Institute (BGI) using the Illumina HiSeq2000 platform. Reads were assembled using Trinity [26] by first combining clean reads with a certain length of overlap to form longer fragments without N into contigs. The reads were then mapped back to contigs with paired-end reads in order to detect contigs from the same transcript as well as to determine the distances between those contigs. Trinity then connected the contigs, using N to represent unknown sequences between every two contigs. From this, scaffolds were made. Paired-end reads were used againfor gap-filling of scaffolds to obtain sequences with the fewest Ns which could not be extended on either end. Suchsequences were defined as unigenes. TGICL [27] at default parameters was used to cluster these unigenes to acquire nonredundant unigenes of the greatest length possible. In the final step, a Blast X alignment was performed between the unigenes and the protein databases of NR, Swiss-Prot, KEGG, and COG. The best aligning results were used to determine the sequence directions of those unigenes. If the results of different databases conflicted with each other, we followed a priority order of NR–Swiss-Prot–KEGG–COG. Orientation and CDS of sequences that have no hits in blast search were predicted using ESTScan [28]. Original transcript sequences (5’->3’) were chosen if their orientations were unable to be determined by above mentioned approaches. Unigene expression levels were calculated using the RPKM method (Reads Per kb per Million reads) [29]. The formula is RPKM= (1000000*C)/(N*L*1000). This formula assigns RPKM(A) to be the expression of gene A, C to be number of reads that uniquely aligned to gene A, N to be total number of reads that uniquely aligned to all genes, and L to be number of bases on gene A. The RPKM method is able to eliminate the influence of different gene length and sequencing discrepancy on the calculation of gene expression. Therefore the calculated gene expression can be directly used for comparing the difference of gene expression among samples.
Data processing
Further isolation of GHs was done by searching the obtained unigene annotation for the keywords:”glycoside, hydrolase, GH, EC: 3.2.1”. These genes were afterwards checked for duplications by blasting gene fragments in NCBI and reverse blasting these results against a local database of obtained unigenes. Blast results lead to the construction of full unigenes without duplications. CLC Main workbench was used for local blast database and for collecting partial genes into full genes.
Peptide Pattern Recognition
Peptide Pattern Recognition (PPR) [24] was used for the identification of GHs on a functional level and of the AA9, AA10 and AA11 families in the Trichodermagenomes as previously described [30]. Briefly, peptide patterns were generated based on all GH and AA9, AA10 and AA11 family proteins in CAZy. For each protein family PPR found the largest group of proteins that contained at least 10 of 70 conserved hexamer peptides as previously described [24]. These conditions (length of the conserved peptides (hexamers), the number of conserved peptides per protein (10) and the total number of conserved peptides per group (70)) were chosen according to previously empirical testing of the parameters that give the best rate of prediction of protein function [30]. Testing included varying peptide lengths from trimers to decamers, number of conserved peptides per protein from 5 to 40 and number of conserved peptides per group from 30 to 200 [22].
The first group of proteins identified was defined as subfamily 1. Next, PPR found the second largest group of proteins, not including any proteins from subfamily 1, defined by the same criteria. This group of proteins was defined as subfamily 2 and so on. PPR continued the analysis until less than five proteins could be grouped in this way. All GH subfamilies containing proteins with a reported enzymatic activity described in CAZy were assigned the same function as the proteinswith a reported enzymatic activity as previously described [22]. All AA9, AA10 and AA11 proteins were assumed to be LPMOs.Gene annotation by finding homology to peptide patterns ,Gene annotation was done as previously described [30] by splitting the genomes into 2000 bases long fragments with 100 bases overlap between fragments and generate all open reading frames in all six frames.For transcriptome analysis, each contig was translated in the three forward reading frames.
Each open reading frame was given a score for each subfamilyspecific peptide lists for each GH and AA family by:
1. Finding all the peptides from the list that were present in the reading frame.
Example: An open reading frame containing the three conserved peptides FGTFHP, GTFHPY and YIKSLD (single-letter amino acid code).
2. Sum the frequency of these peptides to get the subfamily-specific frequency score.
Example: If the peptide FGTFHP is found in 31 % of the proteins in the subfamily, it has a frequency of 0.31. Likewise GTFHPY has a frequency of 0.35 if it is found in 35 % of the proteins in the subfamily and the frequency of YIKSLD is 0.61 if it is found in 61% of the proteins in the subfamily. Thus, an open reading frame containing these three peptides has a subfamily-specific frequency score of 0.31 + 0.35 + 0.61 = 1.27.A hit was considered significant if one of the open reading frames:
1. Included at least three conserved peptides from a subfamily.
2. The sum of the frequency of these peptides was higher than 1.0
3. The conserved peptides covered at least ten amino acids of the ORF.
Example: A protein sequence contains the conserved peptide FGTFHP at residues 11-16, the conserved peptide GTFHPY at residues 12-17 and YIKSLD at residues 66-71. As the peptides FGTFHP and GTFHPY share five amino acids they cover a total of seven amino acids (residues 110-116). Together with the six amino acids covered by YIKSLD it gives a total of 7 + 6 = 13 conserved amino acid residues.
If all three conditions were met a sequence fragment was assigned to the GH or AA family and to the PPR subfamily with the highest subfamily-specific frequency score as previously described [22]. If two fragments were assigned to the same GH or AA family and the distance between them in the original genome sequence was less than 5800 bases, the fragments were considered to be part of the same gene and counted as one hit. Finally, if an enzymatic function had been described for the majority of the proteins in a subfamily the hit was assigned the same function [24].
Phylogeny
Sequences were aligned using ClustalX 2.1 (http://clustalx.software. informer.com/2.1/) with default alignment parameters. Phylogenetic analysis was performed using the neighbor-joining algorithm of ClustalX 2.1 with default parameters (gapped regions were included). Bootstrap analysis (1000 trials) provided a measure of confidence for the detected relationships as described above. The resulting trees were visualized by the program Figtree (http://tree.bio.ed.ac.uk/software/ figtree/)
The choice of substrate is important for the successful production of CAZymes, because different biomasses induce distinct fungal enzyme responses depending on the biomass structure and composition. An initial screening of ten carbohydrate-containing growth substrates was performed to induce the widest and largest production of CAZymes (Supplementary information). T. asperellum produced a wide range of enzymes after only three days incubation. The highest activity was recorded for endo-xylanases and β-glucanases on arabinoxylan, xylan and β-glucan. After three days wheat bran induced the widest range of enzyme activity as well as the largest response in terms of halo size, closely followed by duckweed, Spirodella polyrrhiza. At five days the overall AZCL response increased for all growth substrates except PDA. The widest activity range with the largest halos was observed with wheat bran substrate at seven days (Table 1). Enzymes induced by growth on wheat bran resulted in activity on 8 out of the 13 tested AZCl-substrates, with the highest activity being recorded for xylanases with halo sizes of 4.9cm2 and 4.5cm2 on AZCL-arabinoxylan and AZCL-xylan, respectively. Second, growth on the duckweed Spirodella polyrrhiza induced enzymes that were detected on 7 out of 13 tested AZCL-substrates, but to a lesser degree than wheat bran. This was indicated by significantly less cellulase and 1,4-ß-Dmannanase activity. Enzyme activities when grown on wheat bran were further investigated under different conditions. These included growth durations for five and seven days, temperatures from 200C to 30°C and pH 4, pH 6.5 and pH 8 respectively. Neither the temperature nor the pH of the growth medium had any significant effect on the range of activities measured.
| 7 days | ||||||||||||||||||||
| AZCL/Medium | PDA | WBA | Spirodella polyrrhiza | Lemna minor | Oakmea | wheat bran wheatstraw | Brassica oleracea | Sinapis alba | Cannabies | |||||||||||
| Amylose | 0 | ±0 | 0 | ±0 | 0,0 ±0 | 0 | ±0 | 0 | ±0 | 0,0 ±0 0,0 ±0 | 0,0 ±0 | 0,0 ±0 | 0,0 ±0 | |||||||
| Arabinan | 0 | ±0 | 0 | ±0 | 0,0 ±0 | 0 | ±0 | 0 | ±0 | 0,6 ±0 0,0 ±0 | 0,0 ±0 | 0,0 ±0 | 0,0 ±0 | |||||||
| Arabinoxylan | 0 | ±0 | 38 | ±0 | 3,8 ±0 | 35 | ±0 | 35 | ±0 | 4,9 ±0,2 0,0 ±0 | 3,8 ±0 | 4,2 ±0,1 | 3,8 ±0,1 | |||||||
| Beta-glucan | 0 | ±0 | 8 | ±0 | 11 | ±0 | 6 | ±0 | 9 | ±0 | 31 | ±0 | 15 | ±0 | 20 | ±0 | 15 | ±0 | 20 | ±0 |
| Casein | 0 | ±0 | 6 | ±0 | 8 | ±0 | 8 | ±0 | 6 | ±0 | 11 | ±0 | 8 | ±0 | 13 | ±0 | 11 | ±0 | 15 | ±0 |
| Cellulose | 0 | ±0 | 8 | ±0 | 8 | ±0 | 0 | ±0 | 8 | ±0 | 18 | ±0 | 0 | ±0 | 8 | ±0 | 8 | ±0 | 8 | ±0 |
| Curdlan | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 |
| Galactan | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 |
| Galactomannan | 0 | ±0 | 8 | ±0 | 8 | ±0 | 0 | ±0 | 0 | ±0 | 28 | ±0 | 0 | ±0 | 8 | ±0 | 8 | ±0 | 8 | ±0 |
| Pullulan | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 |
| Rhamnogalactorunan | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 | 0 | ±0 |
| Xylan | 0 | ±0 | 35 | ±0,1 | 45 | ±0,1 | 31 | ±0 | 25 | ±0 | 45 | ±0,2 | 0 | ±0 | 35 | ±0 | 38 | ±0 | 31 | ±0 |
| Xyloglucan | 0 | ±0 | 8 | ±0 | 15 | ±0 | 8 | ±0 | 11 | ±0 | 18 | ±0,1 | 0 | ±0 | 8 | ±0 | 15 | ±0 | 11 | ±0 |
| Sum diameters | 0 | 110 | 133 | 88 | 95 | 207 | 35 | 139 | 145 | 132 |
Table 1: Enzyme activities on different AZCL-substrates after seven days growth on different media,represented by the size of blue AZCL-halos in cm2.
CAZymes in the transcriptome of T. asperellum grown on wheat bran
Duplicates of mycelium from T. asperellum grown on wheat bran at 300C for seven days were harvested and their RNA content was isolated and sequenced according to the description in materials and methods. The sequencing resulted in 12,901,784 reads that were assembled into 32,709 unigenes with an average length of 654nt. The transcriptome sequences were deposited in Transcriptome Shotgun Assembly (TSA) database in Genbank at the NCBI with the accession number SRR1575447. A BLASTp and BLASTx search against the National Center for Biotechnology Information (NCBI)’s protein non-redundant (NR) database showed that 66% of unigenes had significant hits (1e-05). The other 34% of the unigenes had no significant similarity to known proteins or domains in the NCBI database. The latter may contain sequences that mostly covered long untranslated regions of the mRNAs. Among the 32,709 unigenes, growth on wheat bran induced 175 GHs from 48 GH families in T. asperellum (Figure 1 ) and detailed list in the supplementary information Table S4). The majority of the GH families were represented by 1-2 genes, but some families were represented with significantly more genes. The most transcribed family was GH18 with 20 genes, all of which were predicted to be chitinases. 14 genes were transcribed from the diverse GH16 family, of which most were predicted to be endo-1,3- β-glucanases. There were 11 genes each from the GH5 family and GH3 family of which the majority predicted to be endo-β-1,4-glucanases and β-glucosidases, respectively. To determine which genes were transcribed the most, the genes were listed in terms of sequenced raw reads. The most transcribed CWDE was shown to be an amylase represented with 18148 raw reads, which was three times morethan the next enzymes. A 1,3-β-glucanase and a chitinase were the next most transcribed CAZyme with 7743 and 6831 raw reads, respectively.

Figure 1: Number of genes represented per GH family in T. asperellum transcriptome (first column) compared with sequences containing a signal peptide (second column).
Transcriptome derived secretome
To analyze which secreted GHs were transcribed in T. asperellum, the sequenced GHs were analyzed for the presence of a signal peptide. Genes without sequenced 5´ end were evaluated based on whether their homologue from Trichoderma atroviride contains a signal peptide. The transcriptome derived secretome contained 111 enzymes from 37 GH families, which corresponded to 62% of the transcribed GH enzymes covering 75% of the transcribed GH families (supplementary information). The majority of the signal peptide-containing sequences belonged to GH family GH18, GH16 and to some degree GH3 (Figure 1). By contrast, GH76 was represented to a higher degree in the secretome compared with the transcriptome. Listing genes with signal peptide according to number of raw reads showed that the 34 most transcribed genes corresponded to 90% of the raw reads (Table 2).
| GeneID | Rawreads | BGI-annotation | PPR-annotation | EC Number | Family | ||||
|---|---|---|---|---|---|---|---|---|---|
| Unigene396_Lyx4 | 18148 | Amylase | Glu DaY ϭ,ϰ-α-glu Dosidase | 3.2.1.3 | GH15 | ||||
| Unigene562_Lyx4 | 6831 | Chitinase | Chitinase | 3.2.1.14 | GH18 | ||||
| Unigene1796_Lyx4 | 5134 | β-ϭ,ϯ-gluDaŶ | gluDaY eYdo-ϭ,ϯ-β-D-gluDosidase | 3.2.1.39 | GH16 | ||||
| Unigene5138_Lyx4 | 2506 | β-glLJDosidase | * | GH76 | |||||
| Unigene2441_Lyx4 | 2498 | Xylanase | eYdo-ϭ,ϰ-β-dzLJlaYase | 3.2.1.8 | GH11 | ||||
| Unigene2199_Lyx4 | 1702 | β-ϭ,ϯ-edzogluDaYase | gluDaY ϭ,ϯ-β-gluDosidase | 3.2.1.58 | GH55 | ||||
| Unigene5138_Lyx4 | 1507 | α-glLJDosidase | * | GH76 | |||||
| Unigene3977_Lyx4 | 1494 | α-glLJDosidase | *mannan endo-1,6-alpha-mannosidase | * | GH76 | ||||
| Unigene5438_Lyx4 | 1482 | α-glLJĐosidase | * | GH76 | |||||
| Unigene4139_Lyx4 | 1376 | α-L-araďiŶofuraŶosidase | gluĐaŶ ϭ,ϯ-β-gluĐosidase | 3.2.1.58 | GH54 | ||||
| Unigene4884_Lyx4 | 1359 | Chitinase | chitinase | 3.2.1.14 | GH18 | ||||
| Unigene5992_Lyx4 | 1358 | α-glLJĐosidase | *mannan endo-1,6-alpha-mannosidase | * | GH76 | ||||
| Unigene4496_Lyx4 | 1351 | Mannosyl-oligosaccharide glucosidase | mannosyl-oligosaccharide glucosidase | 3.2.1.106 | GH63 | ||||
| Unigene4405_Lyx4 | 1284 | β-gluĐosidase | β-gluĐosidase | 3.2.1.21 | GH03 | ||||
| Unigene2969_Lyx4 | 1158 | α-gluĐosidase | α-gluĐosidase | 3.2.1.20 | GH31 | ||||
| Unigene10314_Lyx4 | 1046 | β-džLJlosidase/α-L-araďiŶofuraŶosidase | džLJlaŶ ϭ,ϰ-β-džLJlosidase | 3.2.1.37 | GH43 | ||||
| Unigene4419_Lyx4 | 1041 | eŶdo-ϭ,ϰ-β-gluĐaŶase | * | GH05 | |||||
| Unigene6388_Lyx4 | 1041 | β-ϭ,ϯ-ϭ,ϰ-gluĐaŶase | * | GH16 | |||||
| Unigene8823_Lyx4 | 939 | α-glLJĐosidase | * | GH76 | |||||
| Unigene5396_Lyx4 | 812 | α-gluĐosidase | α-gluĐosidase | 3.2.1.20 | GH31 | ||||
| Unigene9301_Lyx4 | 743 | EŶdo-ϭ,ϯ;ϰͿ-β-gluĐaŶase | gluĐaŶ eŶdo-ϭ,ϯ-β-D-gluĐosidase | 3.2.1.39 | GH16 | ||||
| Unigene9622_Lyx4 | 639 | Cellobiohydrolase | AA9 | AA9 | GH06 | ||||
| Unigene3741_Lyx4 | 605 | Chitinase | chitinase | 3.2.1.14 | GH18 | ||||
| Unigene4031_Lyx4 | 604 | β-galaĐtosidase | galaĐturaŶ ϭ,ϰ-α-galaĐturoŶidase | 3.2.1.67 | GH28 | ||||
| Unigene9036_Lyx4 | 595 | β-džLJlosidase | džLJlaŶ ϭ,ϰ-β-džLJlosidase | 3.2.1.37 | GH03 | ||||
| Unigene7861_Lyx4 | 589 | β-gluĐosidase | β-gluĐosidase | 3.2.1.21 | GH03 | ||||
| Unigene8089_Lyx4 | 588 | Chitinase | chitinase | 3.2.1.14 | GH18 | ||||
| Unigene14133_Lyx4 | 581 | α-ϭ,Ϯ-ŵaŶŶosidase | ŵaŶŶosLJl-oligosaĐĐharide ϭ,Ϯ-α-ŵaŶŶosidase | 3.2.1.113 | GH47 | ||||
| Unigene7664_Lyx4 | 526 | β-galaĐtosidase | β-galaĐtosidase | 3.2.1.23 | GH02 | ||||
| Unigene6759_Lyx4 | 513 | β-ϭ,ϯ;ϰͿ-eŶdogluĐaŶase | gluĐaŶ eŶdo-ϭ,ϯ-β-D-gluĐosidase | 3.2.1.39 | GH16 | ||||
| Unigene6619_Lyx4 | 494 | Cellobiohydrolase | Đellulose ϭ,ϰ-β-Đelloďiosidase ;reduĐiŶg eŶdͿ | 3.2.1.176 | GH07 | ||||
| Unigene6963_Lyx4 | 467 | Chitinase | chitinase | 3.2.1.14 | GH18 | ||||
| Unigene8432_Lyx4 | 466 | α-L-araďiŶofuraŶosidase | α-N-araďiŶofuraŶosidase | 3.2.1.55 | GH62 | ||||
| Unigene14373_Lyx4 | 421 | α-ϭ,Ϯ-ŵaŶŶosidase | * | GH92 | |||||
Table 2: Transcriptome derived secretome represented by the 34th most transcribed genes containing signal peptide, corresponding to 90% of total secretome in terms of raw reads. Enzymes are listed by number of raw reads. The columns contain BGI-annotation as described in materials and methods. PPR annotation is a result of the predicted function by EC number.
Different substrates induced different CAZyme responses in T. asperellum, based on the initial screening of substrates. To analyze the correlation between growth substrate composition and the secreted CAZymes, the enzymes found in the transcriptome derived secretome were divided into groups of starch degrading, cellulose degrading, hemicellulases, chitinases and unknown based on their predicted function. The transcriptional level based on raw reads of these enzyme groups was compared with the macromolecular composition of wheat bran (Figure 2). A major part of the GHs predicted to be in the secretome were starch degrading enzymes, especially the highly transcribed amylase. The second most produced enzymes were hemicellulases, corresponding to the relatively high degree of arabinoxylan in wheat bran. The least secreted enzymes were cellulases, where the majority of the transcripts were two endo-1,3-β-glucanases from GH16, a family, where similar enzymes has shown also to contain 1,4-β-glucanases activity. Though, endo-1,3-β-glucanases are found to be more involved in anti-mycrobial actions than cellulose degradation [31]. To a lesser transcription degree, two β-glucosidases (GH3) and a cellobiosidase (GH7) were present in the transcriptome derived secretome. Moreover, a relatively large part of the transcriptome derived secretome was chitinases, probably induced to rearrange the cell wall of fungus itself or as a constant anti-microbial response.
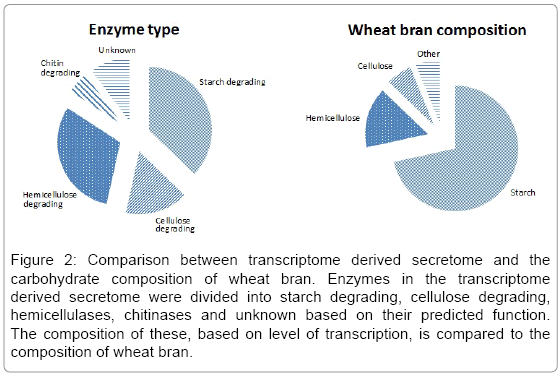
Figure 2: Comparison between transcriptome derived secretome and the carbohydrate composition of wheat bran. Enzymes in the transcriptome derived secretome were divided into starch degrading, cellulose degrading, hemicellulases, chitinases and unknown based on their predicted function. The composition of these, based on level of transcription, is compared to the composition of wheat bran.
Composition of CAZymes across six Trichoderma species
Using a multigene phylogenetic approach, the Trichoderma genus can be divided into four big clades [32]. Clade A “section Trichoderma” consists of among others, T. atroviride, T. hamatum and T. asperellum. Clade B “section Pachybasium” consists of among others T. virens. Clade C “sectionLongibrachiatum” contains T. longibrachiatumand T. reesei. A small clade D consisting of H. aureoviridis. Peptide Pattern Recognition was used to compare the GHs from the genome of five Trichoderma species and the transcriptome from T. asperellumat a functional level (Table 3). Based on PPR analysissignificantly more GHs were identified in the represented species from section Trichodermaand Pachybasium compared to the Longibrochiatum species. T. virens contained a GH profile more like T. asperellum, T. atroviride and T. hamatum rather than T. reesei and T. longibrachiatum. The difference was mostly due to a higher content of hemicellulases and chitinases in the species from Trichoderma and Pachybasium section, but they also contained a higher number of genes within all major enzyme classes (starch degrading, cellulose degrading, hemicellulases, chitinases).
| Enzyme group Annotation, T.longibrochiatum,EC number T.reesei, T.virens , T.atroviride , T.hamatum T.asperellum trans | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Starch | α-aŵLJlase | 3.2.1.1 | 1 | 1 | 2 | 2 | 1 | 2 | ||
| degrading | GluĐaŶ ϭ,ϰ-α-gluĐosidase | 3.2.1.3 | 0 | 1 | 1 | 2 | 2 | 2 | ||
| Dextranase | 3.2.1.11 | 0 | 0 | 1 | 0 | 1 | 0 | |||
| α-glucosidase | 3.2.1.20 | 3 | 4 | 5 | 3 | 6 | 5 | |||
| Cellulose | Cellulase | 3.2.1.4 | 5 | 4 | 7 | 4 | 5 | 4 | ||
| degrading | EŶdo-ϭ,ϯ;ϰͿ-β-gluĐaŶase | 3.2.1.6 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| Oligo-1,6-glucosidase | 3.2.1.10 | 0 | 1 | 2 | 0 | 3 | 1 | |||
| β-glucosidase | 3.2.1.21 | 11 | 11 | 13 | 14 | 15 | 11 | |||
| Cellulose ϭ,ϰ-β-Đelloďiosidase ;ŶoŶ-reduĐiŶg eŶdͿ | 3.2.1.91 | 1 | 1 | 1 | 1 | 1 | 1 | |||
| Cellulose ϭ,ϰ-β-Đelloďiosidase ;reduĐiŶg eŶdͿ | 3.2.1.176 | 1 | 1 | 1 | 1 | 1 | 1 | |||
| β-gluĐaŶs | GluĐaŶ eŶdo-ϭ,ϯ-β-D-gluĐosidase | 3.2.1.39 | 9 | 9 | 10 | 10 | 12 | 9 | ||
| GluĐaŶ ϭ,ϯ-β-gluĐosidase | 3.2.1.58 | 4 | 4 | 9 | 6 | 7 | 4 | |||
| GluĐaŶ eŶdo-ϭ,6-β-gluĐosidase | 3.2.1.75 | 2 | 2 | 2 | 2 | 2 | 1 | |||
| Hemicellulose | EŶdo-ϭ,ϰ-β-džLJlaŶase | 3.2.1.8 | 3 | 4 | 6 | 5 | 7 | 7 | ||
| degrading | Polygalacturonase | 3.2.1.15 | 0 | 1 | 3 | 2 | 2 | 3 | ||
| α-galaĐtosidase | 3.2.1.22 | 4 | 3 | 5 | 6 | 5 | 4 | |||
| β-galaĐtosidase | 3.2.1.23 | 1 | 1 | 2 | 2 | 2 | 1 | |||
| α-ŵaŶŶosidase | 3.2.1.24 | 1 | 1 | 2 | 1 | 1 | 1 | |||
| β-ŵaŶŶosidase | 3.2.1.25 | 5 | 4 | 6 | 6 | 4 | 3 | |||
| XLJlaŶ ϭ,ϰ-β-džLJlosidase | 3.2.1.37 | 3 | 3 | 4 | 3 | 3 | 3 | |||
| α-N-araďiŶofuraŶosidase | 3.2.1.55 | 3 | 1 | 5 | 5 | 6 | 4 | |||
| GalaĐturaŶ ϭ,ϰ-α-galaĐturoŶidase | 3.2.1.67 | 1 | 1 | 3 | 3 | 2 | 1 | |||
| MaŶŶaŶ eŶdo-ϭ,ϰ-β-ŵaŶŶosidase | 3.2.1.78 | 1 | 8 | 1 | 2 | 2 | 1 | |||
| Mannosyl-oligosaccharide glucosidase | 3.2.1.106 | 1 | 2 | 1 | 1 | 1 | 1 | |||
| MaŶŶosLJl-oligosaĐĐharide ϭ,Ϯ-α-ŵaŶŶosidase | 3.2.1.113 | 8 | 0 | 8 | 8 | 8 | 7 | |||
| XLJlaŶ α-ϭ,Ϯ-gluĐuroŶosidase | 3.2.1.131 | 2 | 2 | 3 | 3 | 3 | 2 | |||
| GalaĐtaŶ ϭ,ϯ-β-galaĐtosidase | 3.2.1.145 | 0 | 0 | 0 | 1 | 1 | 1 | |||
| XLJlogluĐaŶ-speĐifiĐ eŶdo-β-ϭ,ϰ-gluĐaŶase | 3.2.1.151 | 1 | 1 | 2 | 2 | 3 | 1 | |||
| GalaĐtaŶ eŶdo-ϭ,6-β-galaĐtosidase | 3.2.1.164 | 1 | 1 | 1 | 1 | 1 | 1 | |||
| α-D-džLJloside džLJlohLJdrolase | 3.2.1.177 | 1 | 1 | 2 | 2 | 2 | 2 | |||
| Chitin | Chitinase | 3.2.1.14 | 14 | 17 | 28 | 23 | 25 | 18 | ||
| degrading | Chitosanase | 3.2.1.132 | 3 | 3 | 4 | 6 | 5 | 1 | ||
| Edžo-ϭ,ϰ-β-D-gluĐosaŵiŶidase | 3.2.1.165 | 1 | 1 | 1 | 1 | 1 | 1 | |||
| Others | AA9 | 2 | 3 | 3 | 2 | 2 | 4 | |||
| Edžo-α-sialidase | 3.2.1.18 | 0 | 0 | 0 | 0 | 0 | 1 | |||
| β-fruĐtofuraŶosidase | 3.2.1.26 | 0 | 0 | 0 | 1 | 3 | 1 | |||
| α-trehalase | 3.2.1.28 | 4 | 4 | 4 | 4 | 4 | 3 | |||
| β-gluĐuroŶidase | 3.2.1.31 | 1 | 2 | 2 | 2 | 2 | 0 | |||
| α-L-rhaŵŶosidase | 3.2.1.40 | 0 | 0 | 1 | 2 | 2 | 1 | |||
| Glucosylceramidase | 3.2.1.45 | 1 | 1 | 1 | 1 | 1 | 1 | |||
| α-N-aĐetLJlgluĐosaŵiŶidase | 3.2.1.50 | 1 | 1 | 0 | 0 | 0 | 0 | |||
| β-N-aĐetLJlhedžosaŵiŶidase | 3.2.1.52 | 3 | 3 | 3 | 3 | 3 | 3 | |||
| GluĐaŶ eŶdo-ϭ,ϯ-α-gluĐosidase | 3.2.1.59 | 3 | 3 | 4 | 2 | 3 | 3 | |||
| ϭ,Ϯ-α-L-fuĐosidase | 3.2.1.63 | 3 | 4 | 4 | 4 | 4 | 1 | |||
| FruĐtaŶ β-fruĐtosidase | 3.2.1.80 | 0 | 0 | 0 | 0 | 1 | 0 | |||
| Total | 110 | 118 | 164 | 150 | 166 | 123 |
Table 3: Comparison of GHs found in publically available genomes of Trichoderma species based on functions predicted by Peptide Pattern Recognition. In the left column are the functions predicted by PPR and bellow the species are the number of genes present in the individual species with this specific function.
PPR analysis identified relatively few starch degrading enzymes, both in terms of functions but also in gene copies, though these were the most transcribed enzyme group based on raw reads. Twice as many hemicellulose degrading as cellulose degrading functions were found across all species, though more were present in T. virens, T. atroviride and T. hamatum compared to T. reesei and T. longibrachiatum. In terms of gene copies, T. reesei and T. longibrachiatum had roughly the same number cellulose acting enzymes as hemicellulose, while T. virens, T. atroviride and T. hamatumcontain around 20% more hemicellulose acting enzymes compared to cellulose acting enzymes. The PPR analysis of GHs at a functional level makes it possible to pinpoint differences in the enzymatic potential of the fungi in terms of specific functions (Table 3). This has revealed for example fewer genes for the Longibrochiatum section within functions with high copy number like chitinase (EC. 3.2.1.14) and β-glucosidase (EC. 3.2.1.21). Further, T. reesei and T. longibrachiatum have lost 9-10 functions, for which the species in section Trichoderma have a low copy number; these include a β-fructofuranosidase, galactan 1,3-β-galactosidase, and α-Lrhamnosidase. The PPR analysis also showed a fructanβ-fructosidase to be present only in T. hamatum and an exo-α-sialidase present only in T. asperellum. Bacterial exo-α-sialidase has been shown to cleave sialic acids from sialyloligosaccharides, gangliosides and glycoproteins. These glycoconjugates are mostly found on surface exposed locations and thought to be among other things involved in cell-to-cell interactions and adhesion [33].
Phylogenetic relationship on a single gene level
Five CWDE-encoding genes were chosen to investigate the evolution on a single-gene level as a further examination of the genetic relationship between the Trichoderma species featured in the study. These genes were chosen based on their high transcription level and presence in the transcriptome derived secretome or because of their significance as the only gene with a specific function. The closest blast hits in NCBI were genes from T. atroviride or T. hamatum and the closest 20-30 hits were sequences from other ascomycete genes. The chosen protein sequences were aligned with all other enzymes from the corresponding GH family or function across all the Trichoderma species using Clustal X. When protein sequences were illustrated in a phylogenetic tree, clades form with related sequences from each Trichodermaand often according to their greater phylogenetic relationship on a species level (supplementary information). This generally form clades with one related sequence from each Trichoderma species, where sequences from the section Trichoderma (T. asperellum, T. hamatum and T. atroviride) exhibit closer relationship relative to sequences from section Longibrochiatum (T. longibrachiatum and T. reesei) and to sequences from the outlier T. virensfrom section Pachybasium. Moreover the genes formed clades with one enzyme from each represented Trichoderma, although on some clades a gene was missing from oneor more Trichoderma. The same trend was seen for the other selected enzymes. To illustrate this point, the phylogenetic tree of endo-1,3- β-D-glucosidasee (EC. 3.2.1.39) is shown (Figure 3).
Figure 3: Phylogenetic tree of predicted glucan endo-1,3-β-D-glucosidases (E.C. 3.2.1.39) present in the featured Trichoderma species.
The production of CAZymes by Trichoderma grown on different substrates illustrated that the choice of substrate is important for the successful production of CAZymes, because biomasses induce a different fungal enzyme response depending on structure and composition. Several Trichoderma species have been successfully cultivated on various lignocellulosic substrates, and their CAZymes analyzed [34,35]. T. asperellum has shown its potential to secrete a range of GHs under appropriate culture conditions [14,22] and when grown on substrates such as cellulose [23]and sugar bagasse [22]. On the latter substrate T. asperellum showed promising results as an enzyme producer exhibiting a higher diversity of hemicellulases and β-glucosidases compared with T. reesei. In addition T. viride exhibited enhanced cellulase production when grown on wheat bran [36] as has T. reesei grown on substrates mixed with wheat bran [37,38]. These studies indicate that several experimental parameters influence enzyme yields, including incubation time, extraction methods, and substrate loading. Other factors that improved cellulase production by T. reesei during solid state fermentation included relative humidity and temperature, continuous light exposure, and aeration [35]. In our study a range of enzyme activities were assayed by AZCL to evaluate the effect of specific growth substrate on the production of CAZymes by T. asperellum. The analysis revealed that substrates induced different enzyme responses from T. asperellum and that more complex sugars such as wheat bran induced a higher and wider activity range than the simple sugars found in e.g. PDA medium, which induced no activity. Beside the sugar complexity, enzyme production also seemed to be dependent on the degree of pretreatment and hence the accessibility to the sugar structure. This may for example be the case with the enzyme response for wheat straw compared to wheat bran; a relatively mild pretreatment of the wheat straw may not have opened up its structure and allowed access to more complex sugars, and this therefore resulted in the production of a relatively simple enzyme cocktail by T. asperellum. Processed wheat bran on the other hand seemed to have the right composition for allowing a broad range of CAZymes to be produced at the same pretreatment level as wheat straw.
CAZymes in the transcriptome of T. asperellum grown on wheat bran
To elucidate the enzymatic response on a transcript level, the transcriptome of T. asperellum was sequenced and glycoside hydrolases were identified the results. When T. asperellum was grown on wheat bran, 175 GHs from 48 GH families were induced. The genome of phylogenetically similar fungi like T. atroviride encodes 213 GHs [39], meaning that around 80% of the GHs present in the genome can be induced by growing T. asperellum on wheat bran. The most represented family was GH18, which was predicted to be chitinases. This response corresponds to other experiments using Trichoderma species that exhibit a high production of chitinases [22,40]. Moreover, several GH16-encoding genes were transcribed. This family contains enzymes with different functions, but all of the expressed GH16s were predicted to be β-glucanases which, like chitinases, are reported to be involved in mycoparasitic activities [40]. Compared with a similar experiment with Trichoderma harzianum [41], GH16 genes are upregulated when growing on sugar bagasse compared with when growing on cellulose and lactose, while a GH16 glucan endo-1,3(4)-β-glucosidase from T. reesei is reported to be highly induced on
Cellulose [42]. A large transcription of GH5 genes was also observed. The genes from GH5 were predicted to contain several functions, including endo-b-1,6-galactanase, endo-1,4-glucanase, β-1,6- glucanase, β-mannanase and xylanase. By contrast, the T. harzianum paper [41] shows that each growth substrate induce its specific GH5 enzyme. This is possiblydue to the simpler structure of substrates in that study compared to wheat bran, and hence fewer enzyme types are needed to bring about degradation. A more in-depth analysis of the enzymes is needed to identify which function is related to which growth substrate. Interestingly, the Trichodermas contain only a few α-glucosidases, making them the smallest group, but α-glucosidases was seen to be the most transcribed enzyme group by T. asperellum, when grown on wheat bran. This may be due to the relatively simple structure of starch, hence needing fewer different enzymes compared to hemicellulose, which due to its more complex structure needs a more complex enzyme cocktail for degradation
Transcriptome derived secretome
To analyze which secreted GHs were transcribed in T. asperellum, the sequenced GHs were analyzed for the presence of a signal peptide. The GH families present in the transcriptomederived secretome were mostly represented by one enzyme per family, but four families exhibited a higher representation: there were five enzymes from GH16, primarily β-glucanases; GH18, primarily chitinases; GH76, α-glucosidases; and GH3, β-glucosidases and xylosidases. Besides being represented by more enzymes than the other families, these were also among the most transcribed genes. Typically, enzyme blends from Trichoderma are low in β-glucosidase activity and are supplemented with β-glucosidases by co-cultivation with other high β-glucosidase-producing fungi to efficiently hydrolyze sugar structure [35]. The enhanced β-glucosidase activity of T. asperellum corresponds to other research where this fungus has been grown on sugar bagasse [22]. But a relatively large part of the transcriptome derived secretome by number of genes was chitinases and β-glucanases. The Trichoderma genus has been reported to produce several β-glucanases and chitinases, which may either be involved in rearranging of the fungus’ own cell wall or as a constant anti-microbial response [40,41,43]. This indicates that wheat bran induces a significant number of anti-microbial enzymes and not just a CAZyme response corresponding with its biomass composition. This might provide the fungi with an advantage in nature, but could represent an energy waste for industrial biomass degradation.
Composition of CAZymes across six Trichoderma species
A drawback of the current classification of GHs in GH families is that each family consists of members with different functions. When a number of enzymes in a GH family have been characterized, the PPR program is able to predict the function of other family members identified only by their sequence. PPR is a non-alignment based method for identifying conserved sequence motifs in biological sequences (for example peptides in proteins) [24]. This provides an opportunity to analyze the fungi in more depth based on GHs and corresponding functions present in the genomes. In the case of family GH76, only one enzyme is characterized, thus PPR is unable to make a reliable prediction. Due to incomplete information about enzymatic properties of the GHs, PPR was only able to predict the function of about 80% of the GHs. This is in the same range as previously found for annotation and functional prediction of 39 fungal genomes [30]. The non-identified GHs were subsequently used in Blast search and their conserved domains analyzed to predict their function. Checking the unidentified GHs limits the possibility of them containing functions described in Table 3. Based on the PPR analysis T. atroviride, T. hamatum and T. virens contained significantly more GHs than T. reesei and T. longibrachiatum. In the case of T. hamatum PPR identified almost 50% more GHs than in T. reesei. This difference was mostly due to a higher number of hemicellulases and chitinases, but T. reesei and T. longibrachiatum also contained fewer enzymes across all major enzyme groups such as starch degrading cellulases and hemicelluloses compared to its mycoparasitic relatives. This was not only the case for enzymes with many copies; based on PPR T. reesei and T. longibrachiatum have lost 9-10 activities. These enzymes belong to a wide range of GH families and are involved in starch, cellulose and hemicellulose degradation. The consequence of this function loss is unclear because T. reesei is regarded as a relatively good lignocellulosic degrader. The overall picture of the PPR analysis corresponds to the current theory of mycoparasitic species, which suggests that the mycotroph-related genes arose in the common ancestor of Trichoderma that had the ancestral mycotrophic life style, and that some of these genes were subsequently lost in saprotrophic T. reesei [43]. This supposedly happened as T. reesei became an efficient saprotroph on dead wood by following wooddegrading fungi into their habitat [44,39]. These findings are also reflected in the phylogenetic comparison that shows a close relation between T. reesei and T. longibrachiatum and a more distant relation to the other species.
Phylogenetic relationship on a single gene level
Phylogenetic trees of the enzymes illustrate clades with similar genes across the species, placing the individual enzymes according to their greater species phylogenetic relationship [32]. This supports the theory that CAZymes in the Trichoderma species have evolved from a common ancestor, followed by an evolutionary process, which resulted in the saprotrophic species especially losing genes [32]. This created smaller clades, where only homologues from some of the Trichoderma species were present. All Trichoderma species contain 6-7 enzymes predicted to be from GH76, and are thus relatively unchanged by the loss of GHs in saprotrophic species. This indicates that these enzymes may have a key role in the degradation of complex biomass, which is also reflected in the high representation of GH76 family in the secretome of T. asperellum when grown on wheat bran. To predict the function of the GH76s with greater accuracy, more members of the family needs to be characterized. One characterized gene from GH76 is a α-1,6-mannanase [45], which shows close similarity to one of the predicted GH76 from T. asperellum. With respect to enzyme discovery, the transcriptome sequencing has elucidated the most transcribed GHs on a specific substrate, which is an indication of which enzymes are important and potentially effective for degradation. PPR on the other hand elucidated the differences between GHs represented in the genomes and revealed specific enzymes and functions for single species within genus Trichoderma.
When T. asperellum was grown on wheat bran, 175 GHs from 48 GH families were transcribed, corresponding to more than 80% of the GHs compared with the closely related T. atroviride. Based on number of raw reads, 90% of enzymes bound for secretion, which are used by the fungi use to degrade biomass, consisted of 35 different enzymes divided on 20 different families, indicating that 40% of the GH families are represented in the secretome. Beside the main carbohydrate degrading enzymes, T. asperellum also transcribed several chitinases and β-glucanases for secretion. This result is suggested to be either for rearrangement of chitin of the fungus itself or a component in antimicrobial action. More detailed insight was gained into the evolutionary differences among Trichoderma spp. by comparing the GHs present in different species. It wasshown that the genetically similar T. atroviride, T. virens, T. hamatum and T. asperellum contain a larger and broader repertoire of GHs compared to the genetically similar T. reesei and T. longibrachiatum. This is additionally supported by the phylogenetic relationship of individual enzymes that form clades of homologues with distances corresponding to the overall phylogenetic relationship between the species. Analysis of enzymes on a functional level instead of on a broader family level allowed the identification of specific functions lost in T. reesei and T. longibrachiatum compared to T. atroviride, T. virens, T. hamatum and T. asperellum, as well as unique functions present in certain species. The latter are important for the discovery of novel GHs involved in plant cell wall degradation.
This work was supported by the Sino-Danish center and Novozymes A/S.