Journal of Sleep Disorders & Therapy
Open Access
ISSN: 2167-0277
ISSN: 2167-0277
Case Report - (2016) Volume 5, Issue 2
Background: The prion diseases are characterized by sleep disruption, with FFI typically characterized also by severe autonomic dysfunction and sympathetic hyperactivity. We report the results of an extensive neurophysiological and autonomic assessment in a CJD patient carrying the D178 mutation with the uncommon homozygosity for valine at codon 129, mutation with long disease duration.
Results: A 47years old female presented with a memory impairment followed by progressive cognitive deficits and ataxia. The clinical picture slowly worsened to a state of akinetic mutism in about 2 years, and the patient died six years after the onset of symptoms. Repeated PSG and long-term actigraphic recordings, showed a peculiar, previously undescribed, pattern characterized by conservation of a rudimental circadian and ultradian rhythm, despite dramatic sleep micro-structure deterioration. We also observed a normal autonomic physiological response to orthostatic challenge and normal dynamic autonomic modulation during wake and sleep. The post-mortem brain pathology study, showed that neuronal loss was substantial in the cerebral cortex, diencephalon and thalami, but not in brainstem nuclei.
Conclusions: We hypothesize that, despite a dramatic neurological picture (i.e. akinetic mutism) and a severe sleep micro-structural alteration, the persistence of an autonomic modulation and the persistence of a rudimental circadian and ultradian oscillation, are related to the relatively conserved anatomo-functional integrity of foundamental neuronal systems in the brainstem.
Keywords: Prion; Sleep; Cardiovascular; Autonomic; Actigraphy; Brainstem
The prion diseases are a family of rare neurodegenerative disorders that result from the accumulation of a misfolded form of the cellular prion protein termed PrPSc. Although the vast majority of cases are sporadic, 10% to 15% are caused by autosomal dominantly inherited mutations in the prion protein gene (PRNP) and include familial Creutzfeldt-Jakob Disease (CJD), Fatal Familial Insomnia (FFI) and Gerstmann-Strãussler-Scheinker disease (GSS).
The D178N PRNP mutation may result in two distinct phenotypes (i.e. CJD or FFI) specified by the polymorphism at codon 129 of the mutant allele, encoding valine in CJD and methionine in FFI [1].
Sleep disruption, initially considered a peculiar hallmark of FFI, has also been described in sporadic and inherited CJD [2-6]. In addition to profound alterations of the sleep-wake cycle, FFI is typically characterized by progressive disappearance of vegetative and hormonal circadian oscillations and severe autonomic dysfunction with sympathetic hyperactivity [5,7].
Here we report the results of an extensive neurophysiological and autonomic assessment carried out during the six year course of the disease at different time points in a CJD patient carrying the D178N-129V mutation. A detailed post-mortem study allowed us to correlate the clinical findings with the neuropathological evidences.
A 47yrs old female presented with a memory impairment followed by progressive cognitive deficits and ataxia. The clinical picture slowly worsened to a state of akinetic mutism in about 2 years, and the patient died for respiratory failure at age 53, six years after the onset of symptoms.
Her father died at age 58 after a two–year history of a similar neurological disorder, and the paternal grandfather died in his 40s after a rapidly progressive dementia. The analysis of PRNP showed that the proband carried the D178N mutation and was homozygous for valine at codon 129. A more detailed clinical history has been already published [6].
The neuropathological examination of the proband [8] showed severe neuronal loss, spongiosis and gliosis in the cerebral cortex, basal ganglia, thalamus and cerebellum and marked myelin loss and tissue rarefaction in the hemispheric white matter, configuring a panencephalopathic form of CJD. Neuronal loss was not evident in brainstem nuclei, including substantia nigra, locus coeruleus, substantia reticularis pontina and inferior olives.
Sleep and autonomic assessment
Two 24-hours full-Polysomnography (PSG) recording, were performed 12 months and 34 months after the onset of the clinical symptoms.
Actigraphic data were collected with a Zero Crossing Mode (ZCM) 16 Hz sample rate and one-minute fixed epoch length. A total of 31 days in two different periods (32 months and 39 months after the onset of symptoms, respectively) were recorded.
Autonomic assessment included the analysis of heart rate variability (HRV), during wake and sleep, and hormonal circadian oscillations.
For HRV analysis, ECG and respiration traces were extracted from PSG recordings (T1 and T2 respectively) during wake (rest and stand position for 10 minutes) and during different sleep stages.
Two samples of 250 beats were selected during rest and stand for dynamic autonomic response to orthostatic challenge.. Autonomic regulation was evaluated by means of spectral analysis of HRV, based on an autoregressive model [8]. HRV identifies three rhythmic oscillations that characterize heart period time series: very low frequency (VLF), low frequency (LF), marker of sympathetic modulation, and high frequency component (HF, synchronous with respiration and marker of vagal modulation).
Sleep and EEG
The results of the two full-PSG recordings were superimposable. The background wake EEG was characterized by symmetrical 8-8.5 Hz rhythm. Irregular sharp wave complexes (mostly triphasic) were observed isolated or with an irregular 1.5-2.5 seconds periodic pattern, and were attenuated by eyes opening. NREM sleep EEG pattern was characterized by monomorphic low amplitude theta and unstable delta activities, with total absence of vertex waves, spindles and K-complex. Absence slow wave sleep and severe reduction of REM sleep were also observed. Only a rudimental wake-sleep rhythm was detectable with a prevalence of wake during day-time and a prevalence of sleep epochs during the night. Ultradian rhythm was undetectable (Figure 1). Movement and respiratory profiles were unremarkable (data not shown).
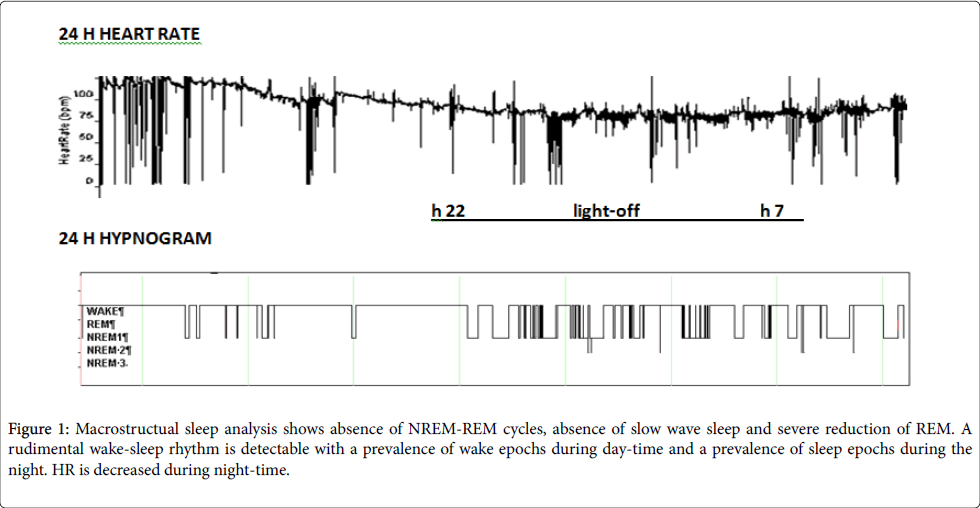
Figure 1: Macrostructual sleep analysis shows absence of NREM-REM cycles, absence of slow wave sleep and severe reduction of REM. A rudimental wake-sleep rhythm is detectable with a prevalence of wake epochs during day-time and a prevalence of sleep epochs during the night. HR is decreased during night-time.
Actigraphic measurements also revealed analogous results: the recording at T1 showed a sleep ratio of 0.66 (Sleep 39.7%, Wake 60.2%) and that of T2 a sleep ratio of 0.63 (Sleep 39%; Wake 61%). A clear prevalence of sleep was observed during night time compared to day time. A peculiar “alternating” pattern was observed during night time (Figure 1).
Autonomic assessment
The orthostatic challenge, performed 54 months after the onset of disease (October 2010) using the rest-stand maneuver, showed an increased heart rate (76 vs 84 bmp) and LFnu, marker of sympathetic modulation (42nu vs 75 nu) during orthostatism, suggesting a normal dynamic autonomic response.
As to sleep analysis, significant differences were observed comparing autonomic control during T1 and T2 and during W and S stages (Figure 2).

Figure 2: A clear prevalence of sleep is observed during night time compared to day time (circadian oscillation). A peculiar “alternating” pattern is observed during night time (ultradian oscillation).
Comparing T2 to T1, both during W and S, a reduction of total variability and a predominant sympathetic modulation were observed. As to sleep analysis, total power - a global index of autonomic variability - increased from W to S at T1 and T2, mainly due to an increased of VLF component. A predominant parasympathetic modulation during sleep and a predominant sympathetic control during wake were present in both recordings (Figure 3).
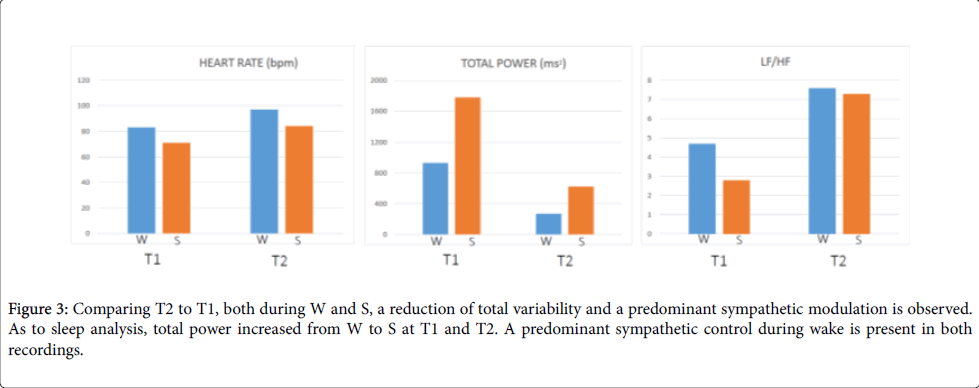
Figure 3: Comparing T2 to T1, both during W and S, a reduction of total variability and a predominant sympathetic modulation is observed. As to sleep analysis, total power increased from W to S at T1 and T2. A predominant sympathetic control during wake is present in both recordings.
We have reported the case of a patient affected by a prion encephalopathy, caused by the D178N mutation with the uncommon homozygosity for valine at codon 129, characterized by an exceptionally long survival (6 years after the diagnosis), compared to the faster disease progression commonly observed in CJD. A collaborative study of human transmissible spongiform encephalopathies carried out from 1993 to 2000, showed indeed a median survival time from less than 6 months in sporadic and familial/ genetic CJD to around 3 years in the GSS disease [7].
In line with previous evidences in CJD, in our case repeated PSG recordings revealed a severe sleep fragmentation with continuous night transitions between wakefulness and light sleep. This is not surprising since sleep disruption and sleep-wake symptoms are commonly recognized, with a remarkable overlap, both in CJD and FFI. The thalamo-cortical neural networks underling sleep consolidation and organization are indeed altered in prion diseases regardless of the specific etiology [3,9].
Despite the severe sleep microstructural alteration, we founded the persistence of a rudimental circadian sleep-wake rhythm, revealed by the prevalence of sleep epochs during night-time as documented by the PSG study. The long-term actigraphic recordings, similarly showed an atypical actigraphic nocturnal cyclic activity (i.e. rest/sleep periods alternated with wake/activity periods) that may be interpreted as the persistence of an ultradian oscillator, in which REM sleep periods are substituted by periods of wake or pseudo-wake state [10,11]. We hypothesize that the persistence of these rudimental circadian and ultradian oscillations, may be related to the, at least partial, sparing of the brainstem regulating network, with particular relevance of the mesopontine (reticular) cholinergic neurons in the peduncolopontine and laterodorsal tegmental nuclei [12]. In our patient, the postmortem brain pathology study [8], showed that neuronal loss was substantial in the cerebral cortex, diencephalon and thalami, but not in brainstem nuclei (substantia nigra, locus coeruleus, substantia reticularis pontina and inferior olives), as described in other cases [13].
The brainstem is considered a critical area also for cardiac autonomic modulation and indeed we observed a normal autonomic physiological response to orthostatic challenge, with an increase of sympathetic and a decrease parasympathetic modulation in orthostatism. Several nuclei in the brainstem are critically involved in the autonomic control of cardiovascular functions, through sympathetic and parasympathetic efferent control on the sinus node and are responsible for the oscillatory components of heart rate variability [14,15]. By the contrary FFI, which usually shows a faster disease progression and a severe autonomic failure, is often characterized by widespread and conspicuous brainstem degeneration [16,17].
In conclusion, we can hypothesize that, despite a dramatic neurological picture (i.e. akinetic mutism) and a severe sleep microstructural alteration, the persistence of an autonomic modulation and the persistence of a rudimental circadian and ultradian oscillator, were related to the relatively conserved anatomo-functional integrity of foundamental neuronal systems in the brainstem. The long survival of our patient carrying the rare VV129-D178 mutation, compared to other CJD forms and FFI, might be the additional consequence.
The work was supported by the Italian Ministry of Health, by the Italian Ministry of Education, Universities and Research (PRIN 2008HC7XR8_002).