Journal of Applied Pharmacy
Open Access
ISSN: 1920-4159
ISSN: 1920-4159
Research Article - (2018) Volume 10, Issue 1
The attempt has been made to compare the aqueous solubility and dissolution characteristics of Aceclofenac (ACE) enhanced by solid dispersion technique using several water soluble carriers PEG 6000, β-Cyclodextrin (β –CD) and Sodium Carboxy methyl cellulose (Na-CMC). Solid dispersions of ACE were prepared with solvent evaporation method. Solid dispersions with carriers were prepared in drug: carrier (1:1, 1:2, 1:3 and 1:4) ratios. The prepared dispersions were evaluated by solubility, in vitro dissolution studies and X-Ray diffraction studies (XRD). The results from XRD analysis showed that ACE might exist in an amorphous state in the solid dispersion. Considerably improved dissolution profile was obtained by PEG ratios (1:2), whereas there was no significant improvement in dissolution of ACE along with β-CD at higher carrier ratios. Na- CMC showed least dissolution improvement among all carriers. The solid dispersion prepared as AC: PEG 6000 (1:2), exhibited the fastest dissolution among all solid dispersions, was formulated into tablets using direct compression method and further compared with immediate release marketed brands of ACE. The results indicated that formulated tablets displayed better dissolution profiles as compared to existing commercial tablets.
<Keywords: Aceclofenac; PEG 6000; β-CD; Na-CMC; Solvent evaporation technique
It has been estimated that nearly 35-40% of drugs suffer from poor aqueous solubility and it affects the absorption of drug from gastrointestinal tract which leads to poor oral bioavailability, high intra and inter subject variability, increase in dose, reduction in therapeutic efficiency and finally failure in formulation development. Various formulation strategies like micronization, solubilization, complexation, dendrimers for drug solubilization, formation of solid solutions/dispersions with hydrophilic carriers, self-micro emulsifying drug delivery systems, spray drying, nano particulate approaches, pro-drug approaches and salt synthesis had been attempted for solubility enhancement. An attractive possibility could be represented by employing simple solid dispersion technique utilizing various hydrophilic carriers. This technique provides a means of reducing particle size to a nearly molecular level, offers a variety of processing and excipients options that allow for flexibility when formulating oral delivery systems of poor water soluble drugs with cost effectiveness and significant dose reduction [1].
Solid dispersions were one of the most successful strategies to improve drug release of poorly soluble drugs. The first description of solid dispersions was from Sekiguchi and Obi in 1961. Formulation of poorly soluble compounds as solid dispersions is one strategy to tackle dissolution-rate-limited oral absorption. Formulation of poorly soluble compounds as solid dispersions might lead to particle size reduction, improved wetting, reduced agglomeration, changes in the physical state of the drug and possibly dispersion on a molecular level, according to the physical state of the solid dispersion [2].
First generation solid dispersions were prepared using crystalline carriers such as urea and sugar, which were the first carriers to be employed in solid dispersion. They have the disadvantage of forming crystalline solid dispersion, which were thermodynamically more stable and did not release the drug as quickly as amorphous ones.
Second generation solid dispersions include amorphous carriers instead of crystalline carriers which are usually polymers. These polymers include synthetic polymers such as povidone (PVP), polyethylene glycols (PEG) and polymethacrylates as well as natural product based polymers such as hydroxypropylmethyl-cellulose (HPMC), ethyl cellulose, and hydroxypropylcellulose or starch derivatives like cyclodextrins.
Recently, it has been shown that the dissolution profile can be improved if the carrier has surface activity or self-emulsifying properties. Therefore, third generation solid dispersions appeared. The use of surfactant such as inulin, inutec SP1, compritol 888 ATO, gelucire 44/14 and poloxamer 407 as carriers was shown to be effective in originating high polymorphic purity and enhanced in vivo bioavailability [3].
Solvent evaporation method: The solvent evaporation method aims to dissolve the drug and carrier simultaneously in a common solvent, followed by the removal of solvent by evaporation. Identification of a common solvent for both drug and carrier can be problematic and complete solvent removal from the product can be a lengthy process. The solvent can be removed by various processes including vacuum drying, heating of the mixture, slow evaporation of the solvent at low temperature, the rotary evaporators, spray drying and freeze drying. Many polymers and drugs that could not be utilized for the melting method due to their high melting points could be used for solvent evaporation method.
Melting method: The melting method involves melting of a physical mixture of drug and carrier to the liquid state followed by cooling until solidification. However, the method is not suitable for thermolabile drugs and incomplete miscibility is observed between the solid drug and molten carrier.
Spray drying process: Spray drying is the process where a solution of drug substance and carrier is evaporated by spraying the solution as fine droplets into a chamber under controlled conditions of heat, humidity and air flow. The drying medium is typically air and the product is then separated after completion of drying.
Hot extrusion method: Hot melt extrusion [HME] is a widely used process in plastic, rubber, and food industry. The process has been useful in the preparation of solid dispersion in a single step. Initially HME is used to prepare solid dispersion to enhance solubility of poorly water soluble drugs. Subsequently, its value in developing controlled release preparation has gained more attraction. The advantages of hot-melt extrusion include lower temperature and shorter residence time of the drug carrier mixture. Typically, physical mixture of drug substance and other ingredient is fed into the heated barrel of extruder at a controlled rate. As the physical mixture is conveyed through heated screws, it is transformed into a fluid like state, which allows intimate and homogeneous mixing by the high shear of extruder screws. The die then shapes the melt in the required form such as granules, pellets, tablets, sheets, sticks or powder.
Supercritical fluid technology: The supercritical fluid exists as a single fluid phase above its critical temperature and critical pressure. Carbon dioxide is the most commonly used supercritical fluid. In GAS or SAS process a mixture of drug and polymer is sprayed through an atomizer into a chamber filled with supercritical fluids. The expansion and extraction of organic solvent into the compressed gas result in lowering the solvent power of organic solvent for drug and polymer leading to precipitation.
Lyophilization (freeze drying): An important advantage of freeze drying is that the drug is subjected to minimal thermal stress during the formation of the SDs. However, the most important advantage is that the risk of phase separation is minimized as soon as the solution is vitrified.
Electrostatic spinning method: Electrostatic spinning method involves the introduction of a liquid into an electric field whereby the liquid is caused to produce fibres. After being drawn from the liquid the fibres harden, which may involve mere cooling, chemical hardening or evaporation of solvent, and then hardened fibres may be collected upon a suitably charged surface. Tubular products comprising polyurethane fibres can be prepared by this electrostatic spinning method. One example of this type of tubular product is a vascular prosthesis, particularly a synthetic blood vessel [4-8].
Hence the main possibilities or improving dissolution according to this analysis are to increase the surface area available for dissolution by decreasing the particle size of the solid compound and/or by optimizing the wetting characteristics of the compound surface, to decrease the boundary layer thickness, to ensure sink conditions for dissolution and, last but definitely not least, to improve the apparent solubility of the drug under physiologically relevant conditions [2].
Drug absorption from the gastrointestinal (GI) tract can be limited by a variety of factors with the most significant contributors being poor aqueous solubility and/or poor membrane permeability of the drug molecule. When delivering an active agent orally, it must first dissolve in gastric and/or intestinal fluids before it can then permeate the membranes of the GI tract to reach systemic circulation. Therefore, a drug with poor aqueous solubility will typically exhibit dissolution rate limited absorption, and a drug with poor membrane permeability will typically exhibit permeation rate limited absorption. Hence, two areas of pharmaceutical research that focus on improving the oral bioavailability of active agents include enhancing solubility and dissolution rate of poorly water-soluble drugs and enhancing permeability of poorly permeable drugs [9-17].
Aceclofenac is a non-steroidal anti-inflammatory drug, which acts specifically on inflammatory sites and thereby decreases the inflammation. It is highly effective as an inflammatory drug for various inflammatory conditions. After oral administration, Aceclofenac is rapidly absorbed and the bioavaibility is almost 100%. Peak Plasma concentrations are reached approximately 1.25 to 3 hours following ingestion. Tmax is delayed with concomitant food intake whereas the degree of absorption is not influenced. It is highly protein bound (>99.7%). Aceclofenac penetrates into the synovial fluid, where the concentration reaches approximately 60% of those in plasma. The volume of distribution is approximately 30L is metabolized into active metabolites in hepatocytes and microsomes. The mean plasma half -life is 4-4.3 hours. Acelofenac circulates mainly as unchanged drug 4-hydroxyaceclofenac is the main metabolite of Aceclofenac. It is also metabolized to number of other metabolites including 5-hydroxyaceclofenac, diclofenac, 4-hydoxyaceclofeac and 5-hydroxydiclofenac. Approximately two-thirds of the administered dose is excreted via the urine, mainly as conjugated hydroxymetabolites. Only 1% of an oral single dose is excreted unchanged. Clearence is estimated to 5 liters per hour. Aceclofenac is probably metabolized via CYP2C9 to the main metabolite 4-hydroxyaceclofenac whose contribution to the clinical activity probably is negligible. Diclofenac and 4-hydroxyaceclofenac have been detected amongst many metabolites [18].
Aceclofenac has major drawback of low aqueous solubility that delays its absorption and has poor oral bioavailability. Practically it is insoluble in water, freely soluble in acetone, soluble in alcohol. It is practically insoluble in water, so the improvement of its dissolution is an important issue for enhancing its bioavailability and therapeutic efficacy. The solution lies in preparing solid dispersion of Aceclofenac with several hydrophilic polymers to increase its aqueous solubility and dissolution [2].
Aceclofenac was obtained as a gift sample from Yarrow Chem products (Batch no-12031216V), Mumbai, India. PEG 6000 (Batch no-0000147880), Sodium CMC (Batch no-13050295V) and β-Cyclodextrin (Batch no-11070915V) were obtained from CDH, Delhi. All the solvents as well as reagents used in the current study are pure and of analytical grades.
Calibration plot
Standard calibration curve of ACE in 0.1 N HCl: 100 mg of ACE was transferred into 100 ml methanol in volumetric flask. 10 ml of the sample was withdrawn from this solution and diluted to 100 ml with 0.1N HCl to form 100 mcg/ml (stock solution) then concentration made by withdrawing 0.5, 1, 1.5, 2, 2.5, 3, 3.5 and 4 ml from stock solution and diluted to 10 ml with 0.1N HCl to make solution of concentration 5 ug/ml, 10 ug/ml, 15 ug/ml, 20 ug/ml, 25 ug/ml, 30 ug/ml, 35 ug/ml and 40 ug/ml. Absorbance was taken at λmax of 271 nm using shimadzu UV/ visible 1700 double beam spectrophotometer and graph was plotted for absorbance versus concentration of ACE.
Standard calibration curve of ACE in pH 6.8 PBS: 100 mg of ACE was transferred into 100 ml methanol in volumetric flask. 10 ml of the sample was withdrawn from this solution and diluted to 100 ml with 6.8 phosphate buffer to form 100 mcg/ml (stock solution) then concentration made by withdrawing 0.5, 1, 1.5, 2, 2.5, 3, 3.5 and 4 ml from stock solution and diluted to 10 ml with phosphate buffer 6.8 to make solution of concentration 5 ug/ml, 10 ug/ml, 15 ug/ml, 20 ug/ml, 25 ug/ml, 30 ug/ml, 35 ug/ml and 40 ug/ml. Absorbance was taken at λmax of 271 nm using shimadzu uv/visible 1700 double beam spectrophotometer and graph was plotted for absorbance versus concentration of ACE.
Determination of melting point
The drug was filled in capillary tube and capillary was placed in the melting point apparatus (model no.1013A, Perfit, India). The temperature required to melt the drug was noted. The same procedure was repeated for three times and mean of three readings was calculated.
Drug-excipients compatibility studies
The infrared absorption spectrum (IR Tracer 100 from Shimadzu, Japan) of ACE was obtained by finely grounding and dispersing the drug in approximately 300 mg finely powdered potassium bromide (KBr). The spectrum of ACE and the corresponding reference standard was recorded in the region of 4000 to 500 cm-1. The FTIR absorption spectrum of ACE exhibits maxima only at the same wavelengths as that of a similar preparation of the corresponding reference standard, thus the FTIR spectrum of the substance being examined should be concordant with the reference spectrum of the drug.
Similarly the drug and polymer were mixed physically in 1:1 ratio and the mixtures were placed in sealed vials for 3 months at room temperature. The pure drug and physical mixture of drug and excipients were scanned by FTIR.
Determination of solubility of drug in different media
Solubility study of ACE was carried out in distilled water, 0.1N HCl and pH 6.8 phosphate buffer solutions. An excess of drug was dissolved in 20 ml media in conical flask and was kept at mechanical shaker for one day followed by filtration of solution. The solutions were then centrifuged for 10 min, supernatant was filtered, suitably diluted and absorbance were recorded at predetermined absorption maxima. All experiments were conducted in triplicate.
Determination of partition coefficient of drug
100 mg of ACE was added in 10 ml of distilled water and was taken in separating funnel along with 10 ml of n-Octanol. It was shacked for 30 minutes followed by separation of aqueous as well as organic layer. 1 ml of aqueous layer was diluted suitably and absorbance was taken at 271 nm. The partition coefficient was calculated from following formula:
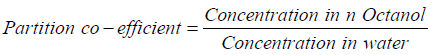
Preparation of solid dispersion
Solid dispersions of ACE were prepared by solvent evaporation method. The drug to polymer ratio used is given in Table 1. An accurate amount of ACE and carrier were taken and dissolved in methanol with continuous stirring. The solvent was removed at 40°C-45°C on hot plate until solid dispersion was obtained. The mass was dried, pulverized and passed through # 44 and was stored in desiccator [4].
| Sl No. | Drug (mg) |
Drug: Carrier ratio | Carriers | ||
|---|---|---|---|---|---|
| PEG 6000 (mg) | β –CD (mg) | Na CMC (mg) | |||
| F1 | 100 | 1:1 | 100 | - | - |
| F2 | 100 | 1:2 | 200 | - | - |
| F3 | 100 | 1:3 | 300 | - | - |
| F4 | 100 | 1:4 | 400 | - | - |
| F5 | 100 | 1:1 | - | 100 | - |
| F6 | 100 | 1:2 | - | 200 | - |
| F7 | 100 | 1:3 | - | 300 | - |
| F8 | 100 | 1:4 | - | 400 | - |
| F9 | 100 | 1:1 | - | - | 100 |
| F10 | 100 | 1:2 | - | - | 200 |
| F11 | 100 | 1:3 | - | - | 300 |
| F12 | 100 | 1:4 | - | - | 400 |
Table 1: Formulation Ingredients of ACE solid dispersions.
Evaluation of solid dispersion
Percent practical yield: Percent practical yield were calculated to know about percent yield or efficiency of any method, thus it helps in selection of appropriate method of production. Solid dispersion were collected and weighed to determine practical yield by following equation:
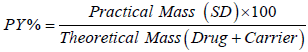
Drug content: The solid dispersion equivalent to 20 mg of drug was taken and dissolved separately in 20 ml of methanol. The solution was kept in rotary shaker for 72 h. The solutions were filtered using 0.45 um membrane filter and were further diluted and assayed by UV spectrophotometer at 274 nm. The actual drug content was calculated by following equation;
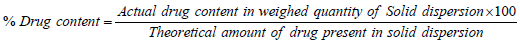
In-vitro Drug release studies: The dissolution studies of solid dispersion were performed using USP II dissolution test apparatus. Volume of media will be 900 ml with a stirring speed of 50 rpm and temperature of medium will be maintained at 37 ± 0.5°C. These conditions were kept constant for all dissolution studies. The drug release study was carried out in 0.1 N HCl for 2 h, later release studies were carried out in pH 6.8 phosphate buffer solutions using dialysis bag. ACE concentrations were determined by UV spectrophotometer at a wavelength of 271 nm. Then the percent cumulative drug release at different time intervals was calculated [5].
Kinetic analysis of drug release: Several theories/kinetics models describe drug dissolution from immediate and modified release dosage forms. There are several models to represent the drug dissolution profiles where f is a function of t (time) related to the amount of drug dissolved from the pharmaceutical dosage system [6]. The kinetics for ACE release from prepared solid dispersion were examined based on the magnitude of correlation coefficients obtained after application of zero order, first order, Hixson Crowell cube root and Higuchi diffusion models.
Zero order kinetics: Drug dissolution from pharmaceutical dosage forms that do not disaggregate and release the drug slowly (assuming that area does not change and no equilibrium conditions are obtained) can be represented by the following equation:
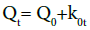
Where, Q0 is the initial amount of the drug in the dosage form, Qt is amount of drug in the dosage form at time t and k0 is zero order rate constant.
First order kinetics: This model has been also used to describe absorption and/or elimination of some drugs describes absorption and/or elimination of some drugs [7] although it is difficult to conceptualize this mechanism in a theoretical basis. The following relation can also express this model:
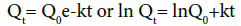
Where, Qt is the amount of the drug released in time t, Q0 is the initial amount of the drug in the solution and k is first order rate constant.
Higuchi model: Higuchi developed several theoretical models to study the release of water soluble and low soluble drugs incorporated in semi-solid and/or solid matrixes. Mathematical expressions were obtained for drug particles dispersed in a uniform matrix behaving as the diffusion media. To study the dissolution from a planar system having a homogeneous matrix, the relation obtained was the following:
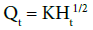
Where, Qt is the amount of the drug released in time t, and KH is Higuchi dissolution constant.
4.8.8 Hixson and Crowell: Hixson and Crowell (1931) recognized that the particle’s regular area is proportional to the cube root of its volume. They derived the equation:
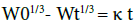
Where W0 is the initial amount of drug in the pharmaceutical dosage form, Wt is the remaining amount of drug in the pharmaceutical dosage form at time t and κ (kappa) is a constant incorporating the surface volume relation. The equation describes the release from systems where there is a change in surface area and diameter of particles or tablets.
X-Ray Diffractometry (XRD): The x-ray diffraction pattern of ACE and carriers, with its solid dispersion were carried out using Philips analytical XRD B.V. (Model PW 3719) and with chromium target and Philips analytical XRD B.N. The conditions were voltage-30Kv; temperature of acquisition room temperature; detector-scintillation counter detector; sample holder-non rotating holder. Solid dispersion powdered was packed in a specimen holder made of aluminium. The powders were passed through a 100 mesh sieve and were placed into the sample holder by the side drift technique. The holder consists of central cavity. In order to prepare a sample for analysis, a glass slide was clipped to the top face of sample holder so as to form a wall. Each powder was filled into the holder gently and used for XRD analysis.
Evaluation of solid dispersion formulated tablets
Thickness: Thickness of the tablets was measured by varnier calipers. Three tablets were selected randomly from all the batches. The diameter of tablets thickness is expressed in mm.
Hardness: Hardness indicates the ability of a tablet to withstand mechanical shocks while handling. Hardness of solid dispersion formulated tablets was determined using hardness tester. Three tablets were randomly picked from each batch and analyzed for hardness. It is expressed in kg/cm2.
Weight variation: From each batch twenty tablets were selected at random and weight was determined. Then the tablets were weighed individually and each weight was compared with an average weight. The variation in the weight is expressed in terms of the % variation.
Friability test: The friability of six tablets was determined by using Roche friabilator. Friability can be determined by the following equation.

In-vitro disintegration test: The tablet disintegration was carried out by placing one tablet in each tube (6 tablets) of the basket and the assembly was suspended in a beaker containing 0.1 N HCl (gastric pH 1.2) and operated without the disc for 120 minutes by maintaining temperature at 37 ± 2°C. The experiment was carried out in triplicate.
Determination of melting point
The reported melting point was 158°C and the observed melting point was found to be 157°C ± 1.54 (n=3), which is nearby to reported value. From the result of determination of melting point, the drug was identified as Aceclofenac.
Fourier Transform Infrared (FT-IR) spectroscopy analysis of drug-excipients compatibility
The Infra-red spectrum of ACE is shown in Figure 1 and the characteristics peaks are depicted in Table 2, which revealed that given drug is Aceclofenac. The FT-IR spectra of physical mixture of drugpolymer are given in Table 3 and represented in Figures 1-7. In the FTIR spectrum of drug, excipients and physical mixture of drug and excipients, all major peaks of drug and excipients are visible in the spectrum. From the above observation it has been concluded that there is no such major shifting of the peaks of the mixtures in comparison to their individual data. Hence the result of the study reveals a good compatibility between drug and polymer.
| Sl. No | Assignments of bands (cm-1) | Reported band positions (cm-1) | Observed band positions (cm-1) |
|---|---|---|---|
| 1 | -N-H(s) | 3500-3180 | 3315.88 |
| 2 | -O-H(s) | 3650-3300 | 3630.92 |
| 3 | -Ar -C-H(s) | 3020-3000 | 3017.29 |
| 4 | -C=O(s) | 1725 | 1720.29 |
| 5 | -C-O-C(s) | 1300 | 1307.56 |
| 6 | -C-Cl(s) | 785-540 | 753.38 |
Table 2: Major band assignments of Aceclofenac.
| Samples | FTIR Peaks | ||||
|---|---|---|---|---|---|
| -OH | -N-H | Ar-CH | -C-Cl | -C=O | |
| Drug | 3630.92 | 3315.88 | 3017.29 | 735.38 | 1720.2 |
| Drug- PEG 6000 | 3321.68 | 3466.12 | 2884.56 | 768.18 | 1815.4 |
| Drug-β –CD | 3320.10 | 3213.45 | 2926.67 | 708.96 | 1717.6 |
| Drug-Na CMC | 3321.34 | 3280.00 | 3145.05 | 750.14 | 1767.0 |
Table 3: Major IR peaks of drug in physical mixture of Drug-Polymers.

Figure 1: FT-IR spectra of Aceclofenac.

Figure 2: FT-IR spectra of β-CD.

Figure 3: FT-IR spectra of physical mixture.

Figure 4: FT-IR spectra of PEG 6000.

Figure 5: FT-IR spectra of physical mixture of Aceclofenac and PEG 6000.

Figure 6: FT-IR spectra of Na-CMC.

Figure 7: FT-IR spectra of physical mixture of Aceclofenac and Na-CMC.
Partition coefficient of drug
The partition coefficient of ACE was found to be 1.56 ± 0.0005. The reported value of partition coefficient is 1.87 [8]. This drug belongs to the BCS class II (low solubility and high permeability), and thus is classified as lipophilic drug.
Percentage practical yield
The % practical yield of ACE: PEG 6000 solid dispersions ranges from 89.45 ± 0.82 to 94.15 ± 0.72 while that of ACE:β –CD and ACE: Na-CMC solid dispersion formulation, it ranges from 82.87 ± 0.87 to 94.76 ± 0.99 and 71.76 ± 0.75 to 83.87 ± 0.34 respectively. The highest among all three drug: carrier solid dispersion was 94.76 ± 0.99 in ACE: β –CD (1:3) formulation. Low coefficient of variance (CV) values (<1.0%) in percentage yield indicates the reproducibility of the technique employed for the preparation of SDs. The % yield of different formulation is shown in Tables 4 and 5.
| Sl. No | Solvents | Solubility (mg/ml) | Reported Value (Arslan A.S., 2010) |
|---|---|---|---|
| 1 | Distilled water | 0.064 ± 0.532 | 0.075 |
| 2 | pH 6.8 PBS | 1.092 ± 0.234 | 1.53 |
| 3 | 0.1N HCl | 0.023 ± 0.086 | 0.015 |
| 4 | Methanol | 2.654 ± 0.65 | 3.876 |
Table 4: Solubility of Drug in different media.
| Percentage Yield* | ||||
|---|---|---|---|---|
| Carrier | Drug: Carrier Ratio | |||
| 1:1 | 1:2 | 1:3 | 1:4 | |
| PEG 6000 | 93.21 ± 0.94 | 91.32 ± 0.81 | 94.15 ± 0.72 | 89.45 ± 0.82 |
| β –CD | 94.76 ± 0.99 | 89.54 ± 0.93 | 91.65 ± 0.64 | 82.87 ± 0.87 |
| Na-CMC | 83.87 ± 0.34 | 79.76 ± 0.24 | 72.87 ± 0.98 | 71.76 ± 0.75 |
Table 5: Percentage yield data of different ACE: Carrier solid dispersions.
Drug content
The drug content of all prepared solid dispersions were found to be in range of 68.87 ± 0.54 to 98.52 ± 0.64. The maximum drug content was found to be 98.52 ± 0.64 in ACE:PEG 6000 (1:3) formulation. Low value for CV (<1.0) indicates uniformity of drug content in the product and suitability of method of preparation. Drug content of all the formulation is shown in Table 6.
| Carrier | Drug: carrier ratio | |||
|---|---|---|---|---|
| 1:1 | 1:2 | 1:3 | 1:4 | |
| PEG 6000 | 97.26 ± 0.28 | 94.34 ± 0.76 | 98.52 ± 0.64 | 97.84 ± 0.98 |
| β–CD | 91.56 ± 0.87 | 84.87 ± 0.76 | 85.87 ± 0.74 | 79.65 ± 0.324 |
| Na-CMC | 81.87 ± 0.65 | 76.87 ± 0.76 | 69.87 ± 0.23 | 68.87 ± 0.54 |
Table 6: Drug content data of different ACE: Carrier solid dispersion.
Solubility studies
The solubility study of all solid dispersion formulation was carried out in 0.1 N HCl and pH 6.8 phosphate buffer solutions. The maximum solubility of 1.28 ± 0.041 mg/ml was found in PEG 6000 based solid dispersion in 1:2 ratios in pH 6.8 phosphate buffer solutions. While in 0.1 N HCl, again ACE: PEG 6000 solid dispersion in 1:2 ratios showed highest solubility among all formulations. The solubility of pure ACE was less as compared to its SDs. The solubility of ACE was increased by using different carriers (PEG 6000, β–CD and Na CMC) which may be attributed to hydrophilic nature of carriers [5]. The solubility profile of all solid dispersion formulation in 0.1 N HCl and pH 6.8 PBS is shown in Tables 7-9. The graphical representation of all formulation is shown in Figures 8-10.
| Solvent | Drug: Carrier Ratio | Solubility (mg/ml) |
|---|---|---|
| 0.1N HCl | 1:1 | 0.066 ± 1.23 |
| 1:2 | 0.089 ± 0.98 | |
| 1:3 | 0.071 ± 1.67 | |
| 1:4 | 0.059 ± 0.95 | |
| 6.8 PBS | 1:1 | 1.15 ± 0.052 |
| 1:2 | 1.28 ± 0.041 | |
| 1:3 | 1.19 ± 0.086 | |
| 1:4 | 1.09 ± 1.07 |
Table 7: Solubility of PEG 6000 Based Solid Dispersion in 0.1N HCl and pH 6.8 PBS.
| Solvent | Drug Carrier Ratio | Solubility (mg/ml) |
|---|---|---|
| 0.1 N HCl | 1:1 | 0.082 ± 0.014 |
| 1:2 | 0.079 ± 0.035 | |
| 1:3 | 0.058 ± 0.083 | |
| 1:4 | 0.061 ± 0.027 | |
| 6.8 PBS | 1:1 | 1.21 ± 0.0167 |
| 1:2 | 1.12 ± 0.065 | |
| 1:3 | 1.05 ± 0.087 | |
| 1:4 | 1.09 ± 0.85 |
Table 8: Solubility of β -CD Based Solid Dispersion in 0.1N HCl and pH 6.8 PBS
| Solvent | Drug Carrier Ratio | Solubility (mg/ml) |
|---|---|---|
| 0.1N HCl | 1:1 | 0.082 ± 0.014 |
| 1:2 | 0.079 ± 0.035 | |
| 1:3 | 0.058 ± 0.083 | |
| 1:4 | 0.061 ± 0.027 | |
| 6.8 PBS | 1:1 | 1.21 ± 0.0167 |
| 1:2 | 1.12 ± 0.065 | |
| 1:3 | 1.05 ± 0.087 | |
| 1:4 | 1.09 ± 0.85 |
Table 9: Solubility of Na -CMC Based Solid Dispersion in 0.1N HCl and pH 6.8 PBS.

Figure 8: Solubility of PEG 6000 based solid dispersion in 0.1N HCl and pH 6.8 PBS.

Figure 9: Solubility of β -CD based solid dispersion in 0.1N HCl and pH 6.8 PBS.

Figure 10: Solubility of Na CMC based solid dispersion in 0.1N HCl and pH 6.8 PBS.
In-vitro drug release studies
In-vitro release profile of all the solid dispersion formulation was examined in 0.1N HCl for 0-2 h and pH 6.8 phosphate buffer solution for 1-1.5 h. In both 0.1N HCl and pH 6.8 phosphate buffer solid dispersions with all drug: carrier ratios exhibited faster dissolution rates than that of pure drug.
Around, 88% of drug was found to be released in 0.1 N HCl (pH 1.2) in PEG 6000 solid dispersion. Once the media changed to buffer of higher pH, more than 95% of drug was released within half an hour (Table 10 and Figure 11).
| Solvent | Drug Carrier Ratio | Solubility (mg/ml) |
|---|---|---|
| 0.1N HCl | 1:1 | 0.045 ± 0.87 |
| 1:2 | 0.068 ± 0.56 | |
| 1:3 | 0.052 ± 0.34 | |
| 1:4 | 0.047 ± 0.98 | |
| 6.8 PBS | 1:1 | 1.12 ± 0.32 |
| 1:2 | 1.07 ± 0.89 | |
| 1:3 | 1.04 ± 0.87 | |
| 1:4 | 1.10 ± 0.67 |
Table 10: In vitro Dissolution Profile of 1:1 Ratio of All Drug: Carrier Solid Dispersions in 0.1 N HCl and Phosphate Buffer pH 6.8.

Figure 11: In vitro dissolution profile of all Drug: carrier (PEG 6000) solid dispersions in 0.1 N HCl and phosphate buffer pH 6.8.
The β –CD solid dispersion also showed more than 80% release in 2 hour in 0.1 N HCl and more than 90% in pH 6.8 phosphate buffer. However, the system prepared with β –CD showed marked improvement in dissolution rate in equal ratio but increment from ratio 1:1 to 1:2 doesn’t result in proportionate enhancement of ACE release (Table 11 and Figure 12).
| % Cumulative Drug Release | ||||
|---|---|---|---|---|
| Dissolution | Time (min) | Drug: carrier ratio (1:2) | ||
| Media | PEG 6000 | β–CD | Na CMC | |
| 0 | 0 | 0 | 0 | |
| 15 | 11.23 ± 0.36 | 10.76 ± 0.69 | 3.45 ± 0.04 | |
| 30 | 17.98 ± 0.34 | 18.87 ± 0.27 | 10.34 ± 0.032 | |
| 45 | 26.09 ± 0.66 | 27.66 ± 0.64 | 15.23 ± 0.45 | |
| 0.1 N HCl | 60 | 39.78 ± 0.54 | 38.64 ± 0.94 | 22.34 ± 0.56 |
| 75 | 51.98 ± 0.23 | 51.56 ± 0.11 | 31.12 ± 0.97 | |
| 90 | 68.54 ± 0.21 | 59.23 ± 0.57 | 39.34 ± 0.30 | |
| 105 | 75 ± 0.89 | 72.87 ± 0.44 | 52.89 ± 0.07 | |
| 120 | 88.32 ± 0.67 | 80.76 ± 0.003 | 62.89 ± 0.59 | |
| 135 | 93.43 ± 0.32 | 84.98 ± 0.34 | 67.65 ± 0.30 | |
| 150 | 97.54 ± 0.45 | 91.89 ± 0.008 | 77.45 ± 0.59 | |
| 6.8 PBS | 165 | 93.99 ± 0.04 | 83.45 ± 0.39 | |
| 180 | 88.78 ± 0.49 | |||
Table 11: In vitro Dissolution Profile of 1:2 Ratio of All Drug: Carrier Solid Dispersions in 0.1 N HCl and Phosphate Buffer, pH 6.

Figure 12: In vitro dissolution profile of all Drug: carrier (β-CD) solid dispersions in 0.1 N HCl and phosphate buffer pH 6.8.
While Na-CMC solid dispersion showed low dissolution rates as compared to other carrier solid dispersion formulation. It showed only 68% release in 0.1N HCl and took 2 h to release 90% of drug in pH 6.8 phospahte buffer (Table 12 and Figure 13).
| % Cumulative Drug Release | ||||
|---|---|---|---|---|
| Dissolution | Time (min) | Drug: carrier ratio (1:3) | ||
| Media | PEG 6000 | β–CD | Na CMC | |
| 0 | 0 | 0 | 0 | |
| 15 | 15.76 ± 0.04 | 12.87 ± 0.04 | 6.87 ± 0.98 | |
| 30 | 23.09 ± 0.98 | 16.87 ± 0.009 | 13.45 ± 0.04 | |
| 45 | 38.67 ± 0.12 | 24.87 ± 0.12 | 24.34 ± 0.003 | |
| 0.1 N HCl | 60 | 51.87 ± 0.56 | 31.11 ± 0.87 | 31.23 ± 0.75 |
| 75 | 63.78 ± 0.77 | 45.76 ± 0.003 | 36.9 ± 0.89 | |
| 90 | 72.44 ± 0.65 | 57.99 ± 0.87 | 41.34 ± 0.20 | |
| 105 | 79.9 ± 0.36 | 61.99 ± 0.04 | 48.23 ± 0.48 | |
| 120 | 85.23 ± 0.91 | 78.76 ± 0.009 | 54.34 ± 0.78 | |
| 135 | 90.12 ± 0.94 | 83.87 ± 0.76 | 62.34 ± 0.44 | |
| 150 | 94.78 ± 0.84 | 89 ± 0.003 | 71.36 ± 0.02 | |
| 6.8 PBS | 165 | 96.34 ± 0.38 | 92.22 ± 0.47 | 84.34 ± 0.49 |
| 180 | 95.32 ± 0.37 | 87.23 ± 0.004 | ||
Table 12: In vitro Dissolution Profile of 1:3 Ratio of All Drug: Carrier Solid Dispersions in 0.1 N HCl and Phosphate Buffer, pH 6.8.

Figure 13: In vitro dissolution profile of all drug: carrier (Sodium-CMC) solid dispersions in 0.1 N HCl and phosphate buffer pH 6.8.
It was found that dissolution rate of solid dispersion from all the formulations was more than 54.56% in 0.1N HCl within 120 min and 97.34% in pH 6.8 phosphate buffer within 1 h. The highest dissolution was observed with PEG 6000 solid dispersion in ratio 1:2 as compared to β-CD and Na-CMC. The in-vitro release was observed in the manner like PEG 6000>β-CD>Na CMC.
The possible reasons attributed for such release behavior from SDs can be suggested as, that the carrier would have formed a hydrophilic diffusion layer around the drug particles altering the surface hydrophobic characteristics of ACE, reducing its particle size, crystalinity, increases the wettability and prevented the drug agglomeration in dissolution medium. The behavior is typically of a carrier which brings about temporary super saturation followed by precipitation of some parts of drug and thus the drug release becomes constant after a specific drug: carrier ratio value [1]. As it can be seen from constant release behavior after 1:2 ratio in all carrier formulations.
Kinetic analysis of drug release
The release of drug from all formulations was observed to follow the first order release kinetics, since the correlation coefficient (r2) for first order was higher in comparison to zero order release. The data was further subjected to Higuchi equation and Hixson Crowell cube root law. A higher correlation, as indicated by (r2) was observed for the Higuchi matrix release kinetics in all the selected formulations suggesting the diffusion as a probable prominent mechanism of drug release [6]. In diffusion, the rate of dissolution of drug particles within the matrix must be much faster than that of the diffusion rate of drug leaving the matrix.
The correlation coefficient and linear equation for all the formulation are computed with the help of DD Solver software. The selection criteria (r2) and the equations best describing the kinetics of in vitro drug release is given in Tables 13-15.
| % Cumulative Drug Release | ||||
|---|---|---|---|---|
| Dissolution | Time (min) | Drug: carrier ratio (1:4) | ||
| Media | PEG 6000 | β–CD | Na-CMC | |
| 0 | 0 | 0 | 0 | |
| 15 | 9.45 ± 0.23 | 8.76 ± 0.29 | 4.08 ± 0.003 | |
| 30 | 16.89 ± 0.45 | 17.98 ± 0.48 | 12.56 ± 0.06 | |
| 45 | 21.45 ± 0.38 | 22.87 ± 0.65 | 26.45 ± 0.59 | |
| 0.1 N HCl | 60 | 28.67 ± 0.67 | 38.55 ± 0.03 | 34.21 ± 0.49 |
| 75 | 39.34 ± 0.73 | 45.59 ± 0.004 | 38.21 ± 0.19 | |
| 90 | 51.89 ± 0.27 | 59.88 ± 0.56 | 43.12 ± 0.93 | |
| 105 | 66.78 ± 0.89 | 65.76 ± 1.45 | 47.34 ± 0.70 | |
| 120 | 74.87 ± 0.38 | 73.09 ± 0.76 | 59.34 ± 0.39 | |
| 135 | 87.65 ± 0.49 | 81.80 ± 0.005 | 67.78 ± 0.59 | |
| 150 | 91.0 ± 0.78 | 87.56 ± 0.98 | 78.23 ± 0.50 | |
| 6.8 PBS | 165 | 96.67 ± 0.58 | 93.34 ± 0.48 | 85.32 ± 0.003 |
| 180 | 95.11 ± 0.57 | 88.21 ± 0.02 | ||
Table 13: In vitro Dissolution Profile of 1:4 Ratio of All Drug: Carrier Solid Dispersions in 0.1 N HCl and Phosphate Buffer, pH 6.8.
| Carrier | Drug: Carrier ratio | Zero order | First order | Higuchi | Hixson Crowell |
|---|---|---|---|---|---|
| PEG 6000 | 1:1 | 0.9479 | 0.9690 | 0.9880 | 0.9567 |
| 1:2 | 0.9842 | 0.9999 | 0.9915 | 0.9742 | |
| 1:3 | 0.9442 | 0.9882 | 0.9715 | 0.9484 | |
| 1:4 | 0.9649 | 0.9842 | 0.9615 | 0.9149 | |
| β –CD | 1:1 | 0.9590 | 0.9942 | 0.9715 | 0.9490 |
| 1:2 | 0.9807 | 0.9842 | 0.9915 | 0.9807 | |
| 1:3 | 0.9860 | 0.9999 | 0.9715 | 0.9799 | |
| 1:4 | 0.9870 | 0.9547 | 0.9615 | 0.9104 | |
| Na-CMC | 1:1 | 0.9293 | 0.9942 | 0.9615 | 0.9393 |
| 1:2 | 0.9322 | 0.9842 | 0.9915 | 0.9722 | |
| 1:3 | 0.9910 | 0.9815 | 0.9715 | 0.9910 | |
| 1:4 | 0.9882 | 0.9875 | 0.9713 | 0.9882 |
Table 14: Drug Release Kinetic Profile of ACE: Carrier SDs.
| S. No. | Optimised Formulation | Formulation code |
|---|---|---|
| 1. | ACE: PEG 6000 (1:2) | F1 |
| 2. | ACE: β –CD (1:1) | F2 |
| 3. | ACE: Na-CMC (1:1) | F3 |
Table 15: Optimised formulations.
X-Ray Diffraction studies
The X- ray diffraction analysis was carried out to confirm the change in the crystalline nature of the drug in solid dispersion and pure form. X- Ray diffraction analysis of ACE and solid dispersion are given in Figures 14-17. The characteristic peaks are situated 00 to 400 (2θ) were used for conformation studies. The drug characteristics peaks were observed at 9, 18, 22 and 28 at 2θ values with intensities of 1100, 3600, 820 and 2500 respectively. The X-ray diffraction pattern of ACE: PEG 6000(1:2) also exhibited well defined peaks at 9, 18 and 25 at 2θ values with intensities of 610, 1700 and 1800 respectively. The diffractogram of ACE: β–CD (1:1) displayed well defined peaks at 10, 18 and 28 at 2θ with intensities 1100, 1400 and 1500 respectively. While that of ACE: Na- CMC all peaks are appearing at higher intensities than other two drugs: carrier system. The peak intensities were reduced, indicating the decrease in the drug crystalinity, which may be responsible for the increased solubility of the solid complex when compared to that of the pure drug.

Figure 14: X-Ray Diffraction pattern of Aceclofenac.

Figure 15: X-Ray Diffraction pattern of ACE: PEG 6000 (1:2).

Figure 16: X-Ray Diffraction pattern of ACE: β –CD (1:1).

Figure 17: X-Ray Diffraction pattern of ACE: Na-CMC (1:1).
Formulation and evaluation of tablets
On the basis of solubility and in-vitro dissolution studies of all solid dispersion formulations, three best formulations were selected from each drug: carrier binary system These optimized solid dispersion were compressed into the tablet form and they were evaluated for various parameters like thickness, hardness, weight variation, friability, in vitro disintegration test and in vitro dissolution testing. The various evaluation parameters of solid dispersion formulated tablets and marketed preparation are given in Table 16. The in-vitro release studies of optimized tableted formulation and marketed preparation is shown in Table 17.
| Evaluations Parameters | Solid Dispersion Formulated Tablets | Marketed Tablet (Acelome) | ||
|---|---|---|---|---|
| F1 | F2 | F3 | MF | |
| Thickness (mm) | 4.05 ± 0.035 | 3.34 ± 0.03 | 3.02 ± 0.015 | 3.66 ± 0.015 |
| Hardness (kg/cm2) | 4.29 ± 0.02 | 3.90 ± 0.015 | 4.74 ± 0.03 | 5.19 ± 0.032 |
| Weight (mg) | 252.33 ± 1.15 | 248.33 ± 0.57 | 252.66 ± 0.57 | 199.6 ± 1.15 |
| Friability (%) | 0.8 | 0.78 | 0.8 | 0.82 |
| Disintegration time (min) | 4.33 ± 1 | 5.45 ± 2.30 | 5.89 ± 0.57 | 11.9 ± 0.52 |
| Drug content (%) | 93.893 ± 0.01 | 95.345 ± 0.02 | 98.817 ± 0.02 | 98.993 ± 0.01 |
Table 16: Evaluation parameter of solid dispersion formulated tablets and marketed tablets.
| Time (min) | % Cumulative Drug Release | |||
|---|---|---|---|---|
| F1 | F2 | F3 | Marketed | |
| 10 | 75.9 ± 0.05 | 27.6 ± 0.58 | 14.6 ± 0.84 | 18.56 ± 0.85 |
| 20 | 83.80 ± 0.004 | 41.34 ± 0.29 | 28.56 ± 0.67 | 32.07 ± 0.34 |
| 30 | 89.04 ± 0.56 | 61.56 ± 0.03 | 39.87 ± 0.01 | 48.11 ± 0.56 |
| 40 | 91.13 ± 1.21 | 67.09 ± 0.45 | 53.12 ± 0.04 | 56.23 ± 0.37 |
| 50 | 93.51 ± 0.04 | 80.01 ± 0.06 | 62.89 ± 0.005 | 71.75 ± 0.58 |
| 60 | 94.75 ± 0.23 | 88.67 ± 0.78 | 71.77 ± 0.15 | 83.19 ± 0.39 |
Table 17: Drug release studies of solid dispersion formulated tablets and marketed tablets.
The formulated tablets were found satisfactory with respect to physical parameters such as thickness, hardness, friability and drug content. The tablets also complied with test for uniformity of weight and disintegration time. The dissolution studies from formulated tablets exhibited almost expected dissolution behavior as that previously obtained from their binary systems, with best dissolution enhancement by F1 (ACE:PEG 6000, 1:2). Commercial brand didn’t provided adequate release in an hour while F1 showed more than 85% release within 30 min. This fact revealed that F1 fulfilled the criteria of at least 85% drug release within 30 min which is set by FDA for immediate release dosage forms.
The similarity factor f2 and difference factor f1 are the important model independent parameters for the mathematical comparison of the dissolution data of different formulations. The values of f2 between 50 to 100 as well as f1 between 0 to 15 showed similarity of the dissolution profiles as per the FDA guidance [5]. The values of f2 and f1 were 54.39 and 27.02 respectively for the formulated tablet F1 indicated that dissolution profile of formulated tablet was not similar to that of the marketed brand.
The preformulation studies revealed that the solubility of AC, a weakly acidic drug (pKa 4-5), depends on pH. AC is highly soluble in basic conditions but relatively soluble in water and acidic pH conditions. The partition coefficient of drug was found to be 1.56 indicating lipophilic nature of drug. The ACE solid dispersion was prepared by solvent evaporation method using PEG 6000, β–CD and Na CMC (weight ratio). The solubility of ACE was enhanced in presence of carriers (PEG 6000, β –CD and Na CMC). The highest solubility was found in ACE: PEG 6000 solid dispersion in 1:2 ratios in both 0.1N HCl and pH 6.8 phosphate buffer solutions. The dissolution rate of ACE from all solid dispersion was significantly higher than that of pure drug.
The general trend indicated that there was increase in dissolution rate for solid dispersion in the following order of PEG 6000>β–CD>Na-CMC. IR studies indicated that no chemical interaction between drug and polymer takes place during preparation of solid dispersion of ACE. The in vitro comparison of three binary system of solid dispersion showed that the highest dissolution was observed with PEG 6000 solid dispersion in ratio 1:2 as compared to β-CD and Na-CMC. Solid dispersion system with all carriers showed reduced crystalinity to a greater extent evidenced by marked reduction in the number as well as the intensity of peaks in XRD of solid dispersion formulations.
The physicochemical characterization at solid state indicated that the enhancing effect of binary systems on dissolution was mainly attributed to the transformation of ACE into the amorphous state as well as improvement of solubility of binary systems in presence of water soluble carriers.
The kinetic analysis of drug release showed the release of drug from formulation in following order: First order>Higuchi order>Zero order>Hixson- Crowell. A higher correlation, as indicated by (r2) was observed for the Higuchi matrix release kinetics in all the selected formulations suggesting the diffusion as a probable prominent mechanism of drug release. The best batch of solid dispersion was selected on the basis of solubility and dissolution studies. ACE: PEG 6000 (1:2), ACE: β–CD (1:1) and ACE: Na-CMC (1:1) were selected as optimized solid dispersion that were compressed into immediate release tablets. Optimized tablets were compared with commercial brand of ACE.
The formulated tablets were found satisfactory with respect to physical parameters such as thickness, hardness, friability and drug content. The tablets also complied with test for uniformity of weight and disintegration time. The dissolution studies from formulated tablets exhibited almost expected dissolution behavior as that previously obtained from their binary systems, with best dissolution enhancement by F1 (ACE:PEG 6000, 1:2). F1 fulfilled the criteria of at least 85% drug release within 30 min which is set by FDA for immediate release dosage forms. The values of f2 and f1 were 54.39 and 27.02 respectively for the formulated tablet F1 indicated that dissolution profile of formulated tablet was not similar to that of the marketed brand.
The authors of the article have no conflict of interest.