Advanced Techniques in Biology & Medicine
Open Access
ISSN: 2379-1764
ISSN: 2379-1764
Research Article - (2015) Volume 3, Issue 2
Vesicular Stomatitis Virus (VSV) is currently being studied as a candidate oncolytic agent due to its ability to induce apoptosis in a variety of cancer cells. Previous studies have shown that Matrix (M) protein mutants of VSV, such as rM51R-M virus, act as selective anti-cancer agents by targeting cancer cells while sparing normal cells. Our goal was to promote the use of VSV for the treatment of cervical cancers. The cervical cancer cell line SiHa has been shown in previous studies to be permissive to infection and killing by VSV. We hypothesized that cervical cancer lines are sensitized to VSV due to blockage of the type-1 interferon (IFN) response by Human Papillomavirus (HPV) oncoproteins. However, our results indicated that SiHa cells retained their ability to respond to type I IFN and were sensitive to killing by both wild-type (wt) and M protein mutant VSV (rM51R-M virus) when infected at a high multiplicity of infection. Another cervical cancer cell line, C4-II, was more resistant than SiHa cells to infection by VSV. To augment killing of cervical cancer cells by VSV, we infected cells in the presence of natural compounds with known anti-cancer activities. Curcumin synergized with VSV to kill both SiHa and C4-II cells, while resveratrol, flavokavain B, Echinacea, and quercetin did not offer an added benefit. In conclusion, our results show that cervical cancer cells exhibit resistance to VSV infection but may be sensitized to VSV-induced oncolysis by the addition of curcumin.
Keywords: Oncolytic virus, Cervical cancer, Type I interferon, Natural products
Vesicular Stomatitis Virus (VSV) is a negative-strand RNA virus of the family Rhabdoviridae. VSV is a pathogen of domestic and farm animals leading to a non-lethal disease characterized by lesions in the mucous membranes of the mouth and nose [1]. However, VSV exhibits low virulence in humans likely due to the induction of an effective immune response leading to the suppression of virus replication and spread [2]. Recently, there has been great interest in developing VSV as an oncolytic agent due to its ability to selectively stimulate apoptosis in infected tumor cells through both intrinsic and extrinsic apoptotic mechanisms [3-5]. It has been proposed that the susceptibility of cancer cells to VSV is due to development of defects in type I Interferon (IFN) antiviral responses in cancer cells during tumorigenesis [6,7]. Furthermore, we and other groups have demonstrated that the selectivity of VSV for tumor cells versus normal cells can be enhanced by using VSV strains that induce type I IFN production in infected cells [6,8-11]. One such recombinant strain is rM51R-M virus which contains an arginine to methionine substitution at position 51 of the M protein sequence. The wild-type (wt) M protein of VSV is responsible for inhibiting host gene expression in infected cells, including genes in the host antiviral immune responses. The M51R M protein mutation renders the virus defective at inhibiting host gene expression and thus, in contrast to its isogenic recombinant wt counterpart (rwt virus); rM51R-M virus stimulates expression of genes involved in host innate immune responses [12]. We and others have shown that M protein mutant viruses are effective oncolytic agents due to their ability to selectively kill cancer cells in vivo without causing disease [3,8,13]. Therefore, these mutant strains represent safer alternatives for cancer therapeutics in humans. However, the efficacy of VSV and its mutant strains in cervical cancers has not been evaluated in great detail.
Cervical cancer is one of the most prevalent cancers in women worldwide and is responsible for thousands of deaths per year [14,15]. Additionally, a high percentage of cervical cancer is associated with infection with high risk strains of human papillomavirus (HPV), HPV- 16 and HPV-18. The E6 and E7 proteins of HPV suppress the functions of tumor suppressors, Rb and p53, leading to uncontrolled cell proliferation [15]. In addition, high-risk HPV types have been shown to deregulate immune-related pathways, such as the type I IFN pathway, to promote oncoviral-mediated carcinogenesis [16]. Therefore, HPVinfected cells with defects in IFN signaling may be sensitive to the oncolytic effect of VSV. However, Le Boeuf and colleagues found that cervical cancer cell lines exhibited variation in sensitivity to infection with VSV strains [17]. Therefore, it is important to determine the basis for differential susceptibilities of cancer cells to oncolytic therapies and identify secondary agents that can be used in combination with VSV to treat VSV-resistant cervical cancer cells.
One strategy to increase the effectiveness of VSV in killing cancer cells is through the use of complementary agents, such as those with cytotoxic or immunomodulatory activity [18]. However, cytotoxic drugs are usually associated with toxicity and negative side effects in patients. Therefore, increased studies are investigating the use of commonly used natural dietary or botanical compounds as adjuncts to various forms of cancer therapies [19]. For our study, we selected several natural products with reported anti-cancer properties: curcumin, resveratrol, quercetin, flavokavain B and Echinacea. These compounds represent safe alternatives to common chemotherapeutic agents and may synergize with oncolytic VSV due to their ability to exert anticancer activity by various mechanisms, including modulation of transcription factors and promotion of apoptosis. Our results indicated that cervical cancer cells displayed resistance to VSV alone, but were sensitized to VSV-induced oncolysis through treatment with curcumin. Therefore, this study represents a novel therapeutic strategy for the treatment of cervical cancers that are resistant to oncolytic therapies alone.
Cell lines and viruses
SiHa and C4-II cervical cancer cells were propagated in DMEM media supplemented with 10% fetal bovine serum (FBS). Both cell lines are adherent, grade II, squamous cell carcinoma cells which originated from the cervix of female patients infected with HPV 16 and 18 strains. Recombinant wild-type (rwt) virus was obtained from an infectious cDNA clone containing wt versions of each of the five VSV genes and their intergenic regions [20]. The rM51R-M virus is isogenic to rwt virus and was obtained by introduction of a methionine to arginine (M51R) mutation into the M gene of VSV in the modified cDNA clone [21]. rwt and rM51R-M viruses expressing GFP (rwt-GFP and rM51R-GFP) were a generous gift from Dr. Doulas Lyles from Wake Forest University Baptist Medical Center (Winston-Salem, NC) and have been described previously [22]. Virus stocks were prepared in BHK cells using methods described previously [23].
Natural products
Resveratrol, flavokavain B and curcumin were obtained from LKT Laboratories, Inc. (St. Paul, MN), while quercetin and Echinacea were purchased from Sigma-Aldrich (St. Louis, MO). Resveratrol was used at dilutions of 25, 50 and 100 μmol; Curcumin at 5 and 10 μmol; and flavokavain B, quercetin and Echinacea at 1, 5, and 25 μg/ml. All drugs were diluted in PBS or cell culture media (DMEM plus 10% FBS).
Measurement of cell viability
Viability of VSV-infected and curcumin, resveratrol, quercetin, Echinacea and flavokavain B-treated cells was measured by MTT assay (MTT Cell Proliferation Kit, Roche Diagnostics). SiHa and C4- II cells grown in 96-well plates were infected with rwt and rM51R-M viruses at multiplicity of infections (MOIs) of 1, 10 and 50 pfu (plaque forming units)/ml. In separate experiments, cells were pre-treated with various concentrations of curcumin, resveratrol, quercetin, Echinacea and flavokavain B for 6 h and infected with rwt or rM51R-M viruses as MOIs of 10 or 50 pfu/cell. At 24 and 48 h post-infection, cells were assayed for viability. All data was normalized to mock-infected cells. Experiments were run in triplicate and were carried out 3-5 times. Data show the mean + standard error.
Response of cells to type I IFN
For determining the response of SiHa cells to type I IFN, cells grown in 96-well plates were treated with 0, 5, 50, and 500 international units/ ml (IU/ml) of Universal type I IFN (PBL Assay Science, Piscataway, NJ). Cells were incubated overnight and challenged with rwt virus at an MOI of 10 pfu/cell for 24, 48 or 72 h. Cell viability was measured by MTT assay.
Virus replication in infected cells
SiHa cells grown in 6 well dishes were infected rwt and M51R viruses engineered to express Green Fluorescent Protein (GFP) from the viral genome (rwt-GFP and rM51R-GFP). Cells were infected at MOIs of 1, 10, and 50 pfu/cell. At 6 and 12 hours post-infection, cells were washed with sterile PBS, trypsinized, and resuspended in 400μL ice-cold PBS. Cells were subjected to flow cytometry analysis. A Beckman-Coulter Cytometrics FC 500 MPL was used to determine the percent of GFP positive cells as an indication of the percent of cells with actively replicating virus, and geometric mean expression as an indication of the degree of virus replication within individual cells.
To determine the susceptibility of cervical cancer cells, SiHa and C4-II, to killing by wt and M protein mutant VSV strains, cells were infected with rwt and rM51R-M viruses at Multiplicities of Infections (MOIs) of 1, 10 and 50 plaque forming units/cell (PFU/cell). A MOI of 1 pfu/cell represents infection of approximately 10% of cells. Therefore, infection at this MOI is a measure of the response of cells to infection with low amounts of VSV particles. In contrast, infection of cells at a high MOI (10 and 50pfu/cell) represents synchronous infection of cells and measures the ability of virus to kill infected cells. At 24, 48 and 72 h post-infection, cell viability was measured and expressed as a percentage of mock-infected cells. Figure 1A shows that SiHa cells were resistant to VSV-induced cell killing when infected with either virus at a MOI of 1pfu/cell since approximately 80% of cells remained viable up to 72 h post-infection. However, cells were more sensitive to killing with rwt virus when infected at MOIs of 10 and 50 pfu/cell. In contrast, C4-II cells remained resistant to killing by both strains of VSV regardless of virus concentration and time.
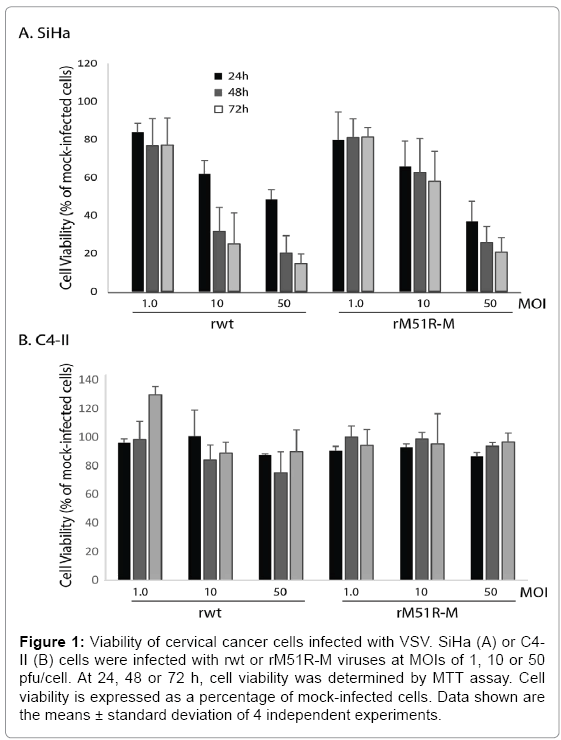
Figure 1: Viability of cervical cancer cells infected with VSV. SiHa (A) or C4- II (B) cells were infected with rwt or rM51R-M viruses at MOIs of 1, 10 or 50 pfu/cell. At 24, 48 or 72 h, cell viability was determined by MTT assay. Cell viability is expressed as a percentage of mock-infected cells. Data shown are the means ± standard deviation of 4 independent experiments.
It is possible that the resistance of cells to killing by VSV is due to residual antiviral responses in infected cells. To test this hypothesis, SiHa cells were pre-treated with various concentrations of type I IFN (0- 500 IU/ml) and challenged with rwt or rM51R-M viruses at a MOI of 10 pfu/cell. At 24, 48 and 72 h post-infection, cell viability was measured by MTT assay. Consistent with data shown in Figure 1, SiHa cells were susceptible to killing by rwt virus when infected at a MOI of 10 pfu/cell such that cell viability decreased to 10% of mock-infected cells by 72 h post-infection (Figure 2). However, cells were more resistant to killing by rM51R-M virus. Upon pre-treatment of SiHa cells with type I IFN at concentrations of 5, 50 and 500 IU/ml, cells developed resistance to VSV-induced cell killing. These results indicate that SiHa cells retain intact antiviral responses that may protect them from infection with viruses. Similar experiments were carried out in C4-II cells and cells remained completely resistant to both rwt and rM51R-M viruses (data not shown).
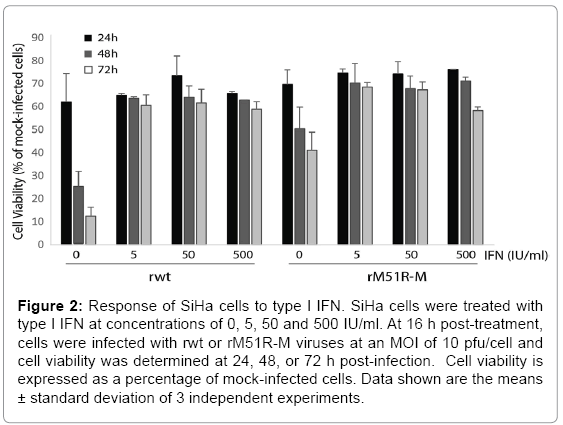
Figure 2: Response of SiHa cells to type I IFN. SiHa cells were treated with type I IFN at concentrations of 0, 5, 50 and 500 IU/ml. At 16 h post-treatment, cells were infected with rwt or rM51R-M viruses at an MOI of 10 pfu/cell and cell viability was determined at 24, 48, or 72 h post-infection. Cell viability is expressed as a percentage of mock-infected cells. Data shown are the means ± standard deviation of 3 independent experiments.
The ability of SiHa cells to mount an antiviral response may result in the attenuation of virus replication in infected cells. To determine the ability of VSV to replicate in infected SiHa cells, cells were infected with rwt-GFP and rM51R-GFP viruses for 6 and 12 h. Cells were harvested and subjected to flow cytometry to determine the percent of GFP positive cells as an indication of the percentage of cells with actively replicating virus (Figure 3A) and the geometric mean fluorescence as an indication of the degree of replication within infected cells (Figure 3B). We observed a dosage-dependent increase in GFP positive cells and geometric mean fluorescence in cells infected with both rwt and rM51R-M viruses (Figure 3A) which is consistent with the cell viability results in Figure 1. Interestingly, a higher percentage of cells infected with rM51R-M virus were GFP positive as compared to rwt virus. This indicates that cells were more permissive to infection with rM51R-M virus than rwt virus, but this did not enhance the downstream ability of rM51R-M virus to kill infected cells.
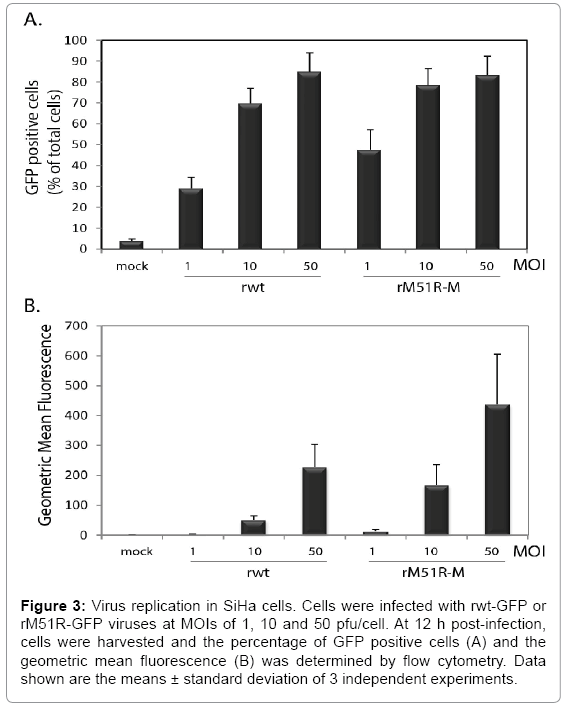
Figure 3: Virus replication in SiHa cells. Cells were infected with rwt-GFP or rM51R-GFP viruses at MOIs of 1, 10 and 50 pfu/cell. At 12 h post-infection, cells were harvested and the percentage of GFP positive cells (A) and the geometric mean fluorescence (B) was determined by flow cytometry. Data shown are the means ± standard deviation of 3 independent experiments.
Our data thus far indicates that cervical cancer cells are relatively resistant to killing by oncolytic VSV. In order to enhance the ability of VSV to kill infected cancer cells, we treated SiHa and C4-II cells with natural products with known anti-cancer activities. Figure 4 shows results from experiments in which we pre-treated cells with 5 and 10μmol curcumin followed by infection with VSV. Rwt and rM51R-M viruses decreased the viability of SiHa cells to approximately 50-60% of mock-infected cells by 72 h post-infection (Figures 4A and 4B). In addition, SiHa cells were resistant to killing by 5 μmol curcumin alone. However, when SiHa cells were pretreated with 5 μmol curcumin and infected with rwt or rM51R-M viruses, there was a significant decrease in cell viability at 48 and 72 h post-infection, as compared to virus infection or curcumin treatment alone. We observed a similar result in C4-II cells when cells were pre-treated with 10 μmol curcumin (Figures 4C and 4D). These data indicate that curcumin synergizes with VSV to kill cervical cancer cells.
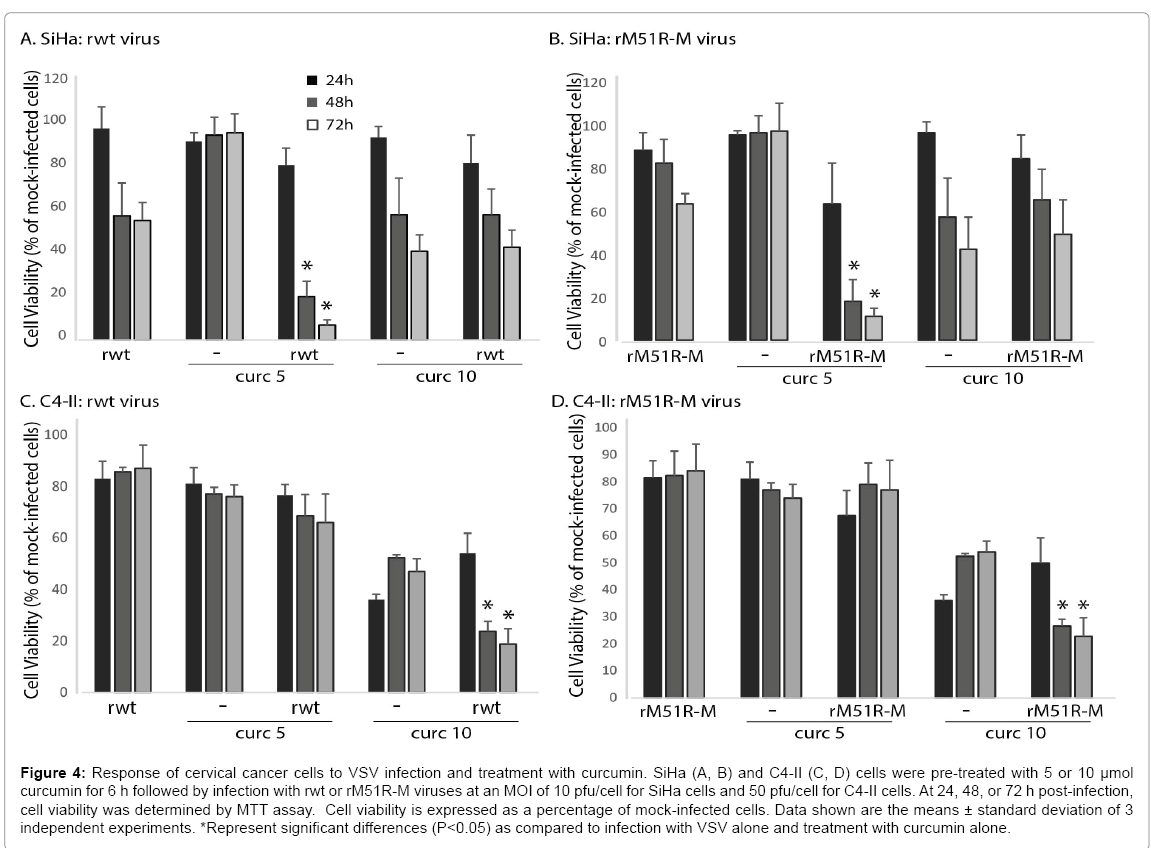
Figure 4: Response of cervical cancer cells to VSV infection and treatment with curcumin. SiHa (A, B) and C4-II (C, D) cells were pre-treated with 5 or 10 μmol curcumin for 6 h followed by infection with rwt or rM51R-M viruses at an MOI of 10 pfu/cell for SiHa cells and 50 pfu/cell for C4-II cells. At 24, 48, or 72 h post-infection, cell viability was determined by MTT assay. Cell viability is expressed as a percentage of mock-infected cells. Data shown are the means ± standard deviation of 3 independent experiments. *Represent significant differences (P<0.05) as compared to infection with VSV alone and treatment with curcumin alone.
To determine if additional natural products are capable of promoting VSV-induced oncolysis, cells were pretreated with various concentrations of quercetin, Echinacea, flavokavain B and resveratrol and infected with rwt or rM51R-M viruses. Pre-treatment with Echinacea significantly sensitized SiHa cells to killing by VSV by 48 h post-infection (Figure 5). Similar results were obtained when SiHa cells were pretreated with 5 μg/ml of flavokavain B and infected with rwt virus for 24 h (data not shown). In contrast, the natural products did not promote killing of C4-II cells by VSV. These data suggest that cervical cancer cells similar to SiHa cells may benefit from the addition of natural compounds prior to oncolytic therapies.
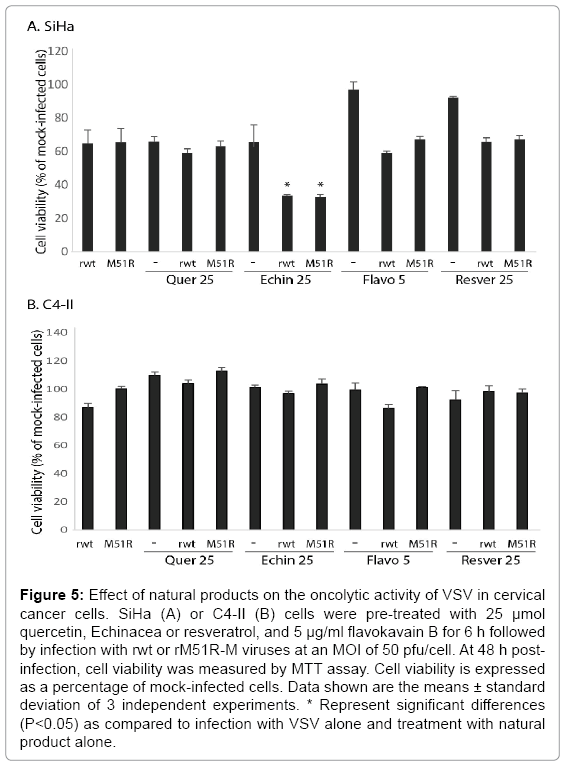
Figure 5: Effect of natural products on the oncolytic activity of VSV in cervical cancer cells. SiHa (A) or C4-II (B) cells were pre-treated with 25 μmol quercetin, Echinacea or resveratrol, and 5 μg/ml flavokavain B for 6 h followed by infection with rwt or rM51R-M viruses at an MOI of 50 pfu/cell. At 48 h postinfection, cell viability was measured by MTT assay. Cell viability is expressed as a percentage of mock-infected cells. Data shown are the means ± standard deviation of 3 independent experiments. * Represent significant differences (P<0.05) as compared to infection with VSV alone and treatment with natural product alone.
The results from this study indicate that SiHa and C4-II cervical cancer cell remain relatively resistant to infection and killing by VSV. The results of this work contradict those seen in previous studies where investigators observed that several cervical cancer cell lines, including CaSki, SiHa, HeLa and ME180, were sensitive to infection and killing by VSV [17]. However, these investigators did not evaluate the C4-II cervical cancer cell line and used VSV strains that differed from ours. Nevertheless, the sensitivity of cervical cancer cells to virus infections is attributed to the functions of the E6 and E7 genes of HPV. In addition to targeting tumor suppressor proteins to promote malignant conversion, they have more recently been shown to inhibit the antiviral IFN response in infected cells. E6 oncoprotein associates with tyrosine kinase 2, Tyk2, prevent its interaction with the type I IFN receptor [24]. This interaction leads to the downstream repression of the Jak- STAT IFN response pathway. More recently, studies have shown that E7 oncoprotein promotes the formation of an inhibitory transcriptional complex that suppresses the antiviral response by downregulating the expression of toll-like receptor 9 (TLR9) [16]. Therefore, it is interesting that in our hands we observed that SiHa cells retained their ability to respond to type I IFN (Figure 2). The resistance of our cell lines to VSVinduced oncolysis may be due to their ability to mount an effective antiviral response against VSV despite the reduced antiviral activity associated with transformation by HPV. Furthermore, since C4-II cells were more resistant to VSV-induced killing than SiHa cells, it is possible that C4-II cells may constitutively express antiviral gene products rendering them inherently resistant to oncolysis by VSV.
Previous studies have shown that the M protein mutant virus, rM51R-M virus, offers great potential as an oncolytic virus for antitumor therapies due to its ability to directly kill tumor cells, while protecting normal cells [6,8]. Additionally, because this virus is a strong inducer of dendritic cell (DC) maturation, it offers promise for initiating tumorspecific T cell responses [25,26]. The M protein of VSV plays multiple roles in the life-cycle of the virus. It is involved in virus assembly as well as in the inhibition of host gene expression in infected cells [27-29]. The overall suppression of host gene expression by VSV promotes viral survival by decreasing expression of genes in the type I IFN antiviral response [12]. The rM51R-M mutant of VSV induces IFN production in infected cells due to its defect in inhibiting host gene expression [12,28]. Therefore, in our study, it was not surprising to observe that rwt virus was more successful than rM51R-M virus at killing SiHa cells. Furthermore, rM51R-M virus replicated more efficiently in SiHa cells than rwt virus (Figure 3). This result is consistent with earlier data in other cell types showing that the M51R protein mutation does not affect the virus assembly functions of M protein and that the rM51R-M virus produces slightly higher virus yields than rwt virus [12,28].
Our results demonstrated that curcumin has the potential to synergize with oncolytic VSV for the treatment of VSV-resistant cervical cancers. Curcumin, an active constituent of turmeric, has been shown to possess anti-oxidant, anti-inflammatory and anti-cancerous properties [30,31]. The anti-cancer properties of curcumin are mediated through its inhibition of several cell signaling pathways [31-33]. Curcumin modulates the activity of transcription factors, growth factors, enzymes and kinases critical for cancer cell survival. Cervical cancer cell progression is associated with constitutively activated Nuclear Factor kappa B (NF-kB) [34]. NF-kB is a transcription factor whose aberrant constitutive activation is involved in oncogenesis and survival in human tumors of diverse tissue origin [35]. In addition, NF-kB also plays an important role in regulating the cellular response to IFNs [36]. Studies have shown that curcumin inhibits the NF-kB signaling pathway in various cancer cells [31,33]. Therefore, pre-treatment of cells with curcumin may further suppress the antiviral response in cervical cancer cells to promote virus infection. Interestingly, the concentration of curcumin that was effective at amplifying VSV-induced oncolysis varied depending on the cell line. It is possible that at higher concentrations in some cell lines, such as SiHa cells, curcumin damages the virus particle or inhibits its ability to effectively infect and replicate in cells. However, at lower concentrations, curcumin may suppress the antiviral response in SiHa cells allowing VSV to replicate to higher levels and promote cell killing. Future studies will focus on the mechanism by which curcumin synergizes with oncolytic VSV in different cervical cancer cell lines.
In addition to curcumin, Echinacea also promoted the ability of VSV to kill SiHa cells. Echinacea has been shown to modulate the expression of multiple transcription factors, including NF-kB, AP-1, AP-2 and numerous STATs [37]. Therefore, it may sensitize certain cervical cancer cells to viral infections by modulating immune-related or apoptotic responses. However, it is unlikely that the modulation of a single transcription factor by curcumin or Echinacea is responsible for the synergy we observed. Flavokavain B, quercetin and resveratrol also have been reported to exert anti-cancer activities through the inactivation of NF-kB and Akt, as well as through reduced expression of anti-apoptotic factors, including Bcl-2, Bcl-xl and XIAP [38,39]. Therefore, additional pathways, as well as the combination of pathways, must be studied in order to understand the underlying mechanisms by which certain compounds promote VSV-induced oncolysis.