Endocrinology & Metabolic Syndrome
Open Access
ISSN: 2161-1017
ISSN: 2161-1017
Review Article - (2018) Volume 7, Issue 5
Obesity is currently considered as the 21st century epidemic. The accelerated increase in prevalence and mortality due to cardiovascular diseases establishes a historical precedent as a global public health problem. The increased incidence of obesity and obesity-associated chronic diseases (coronary heart disease, cancer, diabetes), together with the frequent finding of these conditions in the clinic, urgently call for studies aiming to identify possible pathophysiologic connections among these conditions.
Obesity is often seen only as an imbalance between caloric intake and energy expenditure. On the other hand, numerous neuroendocrine factors are responsible for the regulation of energy metabolism. In addition, body metabolism is also affected by the autocrine, paracrine, and endocrine activity of organokines. Thus, the understanding signaling pathways, action and regulation of organokines could lead to a comprehensive approach to obesity, which in turn may unravel new indicators of adiposopathy, that are not necessarily associated solely to body weight or evident excess of fatty tissue.
Herein we propose a pathophysiological model, which we refer to as the triumvirate of adiposopathy, involving the alteration of organokine balance (myokines, hepatokines, and adipokines) and that takes into account signaling pathways that are common to pro-inflammatory states such as insulin resistance and endothelial damage, emphasizing in adiposopathy and obesity, aiming to achieve the early identification of cardiometabolic risk, and thus positively impact the risk of morbimortality associated with adiposity.
Keywords: Obesity; Fatty tissue; Hepatokines; Myokines; Adipokines
Obesity is a worldwide public health issue whose prevalence is on the rise, along with the associated morbidity and mortality risk of type-2 diabetes mellitus and cardiovascular disease [1,2]. In Colombia, according to the National Nutrition Status Survey of 2015 (ENSIN2015), obesity affects 56% of the adult population [3]. In developed countries, such as the United States (USA) or Canada, 1 in every 6 children present excess weight, and up to 30% of the population younger than age 15 years are obese [4].
Despite the significant prevalence of obesity, it is therefore puzzling that this endocrine disease is not clearly identified by health workers, and is usually referred to as an alteration of the Body Mass Index (BMI). Similarly, the term metabolic syndrome, which has been used for many years, does not represent the pathophysiology of a group of metabolic disorders. Moreover, the clinical criteria for the diagnosis of metabolic syndrome were based on data from studies in the United States and Europe, and may thus not be applicable to other populations such as Latino or Asian. Furthermore, because the diagnosis of metabolic syndrome alone did not seem to be a better predictor of future metabolic disease than the evaluation of its individual components, the National Cholesterol Education Program’s Adult Treatment Panel III report (ATP-III), and the International Diabetes Federation validated the importance of pathogenic adipose tissue by designating an increase in waist circumference (central obesity) as the only necessary criteria of a total of 5 for the diagnosis of the metabolic syndrome, which must then be accompanied by other metabolic abnormalities [5].
What has caused so many changes in the medical nomenclature? Historically, chronic diseases have been treated as independent entities, and thus multiple common factors that may unite them may have been overlooked. In addition, the possibility that different chronic diseases are actually interconnected is not taken into account under this historic perspective, even when this interrelation may explain the multiple associations of different diseases observed in a same patient, as is the case for the widely reported association between obesity, diabetes, hypertension, metabolic syndrome, fatty liver disease, insulin resistance, prediabetes, sarcopenia, among others.
Obesity is often seen only as an imbalance between caloric intake and energy expenditure. Numerous neuroendocrine factors are responsible for the regulation of energy metabolism, and body metabolism is also affected by the autocrine, paracrine, and endocrine activity of organokines, which are proteins produced and secreted by their respective tissues (e.g., adipokines, myokines, hepatokines, cardiomiokines, etc). Thus, the understanding of the relationship between different organokines may provide the answer to the abovementioned question [6].
The proper identification and understanding of organokine pathways (axis), their action on target organs, and the effects of their over-expression or inhibition, allow us to hypothesize that by taking into account the currently available knowledge involving multiple organs, different disorders may be grouped as having a common origin.
Beyond the increase in body weight, abdominal obesity has been previously shown to be a strong and independent predictor of morbidity and mortality, as evidenced by the association between accumulation of adipose tissue, preferential hepatic accumulation, and insulin resistance [7-9]. In its early stages, excess of adipose tissue is characterized by increased lipolysis, free fatty acid release, and altered adipokine production that is secondary to tissue dysfunction due to inflammation or adiposopathy [10]. In this scenario, a bidirectional communication between hepatokines and adipokines is established.
On the other hand, the role of myokines, which mediate muscle growth and enable communication with other organs such as adipose tissue, liver, and pancreas, is well understood [11]. The action of exercise-induced myokines is important for the prevention and treatment of diabetes mellitus type 2, as they play a role in regulation of glucose transporter type-4 (GLUT-4) receptor expression. In addition, myokines, and their mechanism of action, may explain the correlation between sedentary lifestyle and risk of metabolic disorders including obesity, diabetes mellitus type 2, and cardiovascular diseases [12-15]. In agreement with those observations, previous studies assessing the relationship between sedentary behaviour and increased adiposity have evidenced that the ratio of myokines is less than that of adipokines, which in turn increases the metabolic risk of chronic diseases such as diabetes mellitus type 2 [16,17].
The consolidation of all the networks associated with adiposopathy may allow us to broaden the clinical perception of the patient with high body fat composition, and take into account its association with different organ comorbidities, such as hepatic dysfunction [18]. Therefore, in line with this vision, we propose a pathophysiological model that integrates networks of the main organokines associated with adiposopathy, beyond BMI, and is referred to as the CLARos’ model (Rosero et. al, Las Américas Clinic model).
The CLARos’ model acknowledges the homeostatic alteration between organs that make up the "triumvirate of adiposopathy” concept, which summarizes the pathogenic influence of myokines, hepatokines, and adipokines that can lead to inflammation by increasing free radicals that trigger the adiposopathy (Figure 1). The role of the main cytokines involved in adiposopathy and the proposed model of the pathophysiology of adiposopathy are described below.
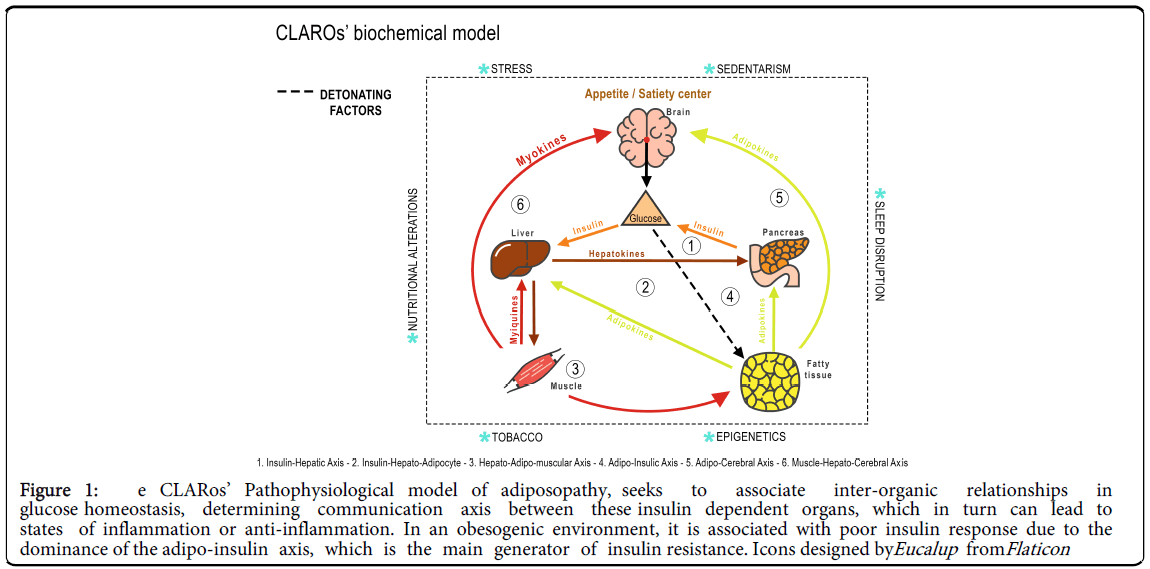
Figure 1: e CLARos’ Pathophysiological model of adiposopathy, seeks to associate inter-organic relationships in glucose homeostasis, determining communication axis between these insulin dependent organs, which in turn can lead to states of inflammation or anti-inflammation. In an obesogenic environment, it is associated with poor insulin response due to the dominance of the adipo-insulin axis, which is the main generator of insulin resistance. Icons designed byE ucalup fromF laticon
Myokines
Skeletal muscle is one of the largest organs in the human body, contributing 40% of the total body weight. Muscle contraction produces and releases a variety of cytokines and other peptides, called myokines [11]. Once secreted, myokines act in autocrine, paracrine, and endocrine fashions, thus being associated with different pathways and organs far from the production site such as visceral fatty tissue, bone, liver and central nervous system [19].
To date, some of the most studied myokines in the context of control and prevention of chronic non-communicable diseases and cardiovascular risk are IL-6, IL-8, IL-15, brain derived neurotropic factor (BDNF), monocyte chemotactic protein 1 (MCP-1), and myostatin [20]. Interleukin-6 (IL-6) is an innate pleiotropic cytokine, produced by hepatocytes, T cells, macrophages, and smooth and striated muscle cells, which regulates the response of the acute phase, inflammation, and hematopoiesis [21]. Its function ranges from stimulating lipolysis and cortisol to having an effect on glucose control. Additionally, it plays a key endocrine role by increasing synthesis of hepatic glucose during physical activity, and increasing lipolysis in adipose tissue [11].
Interleukin-15
(IL-15) is synthesized in skeletal muscle, and its anabolic effect and role as growth factor have been previously shown to promote increase of muscle protein and reduced muscle degradation. Simultaneously, IL-15 has been associated with muscular hypertrophy, optimization of glucose uptake, and to stimulate lipid oxidation in skeletal muscle [22,23]. On the other hand, it has an important role in lipid metabolism, growth and differentiation of B and T lymphocytes, natural killer cells, macrophages and monocytes [11-24]. Neurotropic factor derived from the brain (BDNF) plays an important role in learning and memory, as well as in growth and maintenance of neurons and their synapses [25,26], by acting on adipose tissue to increase its use as an energy source. A significant decrease in this myokine has been observed in obesity and diabetes mellitus type 2 [27].
Monocyte chemotactic protein 1
(MCP-1) is a collaborative contraction-induced myokine, with similar activity to IL-6, and IL-8. It has been found in blood, in longterm exercises such as marathons [28].
Apelin
Apelin is a recently described vasoactive peptide that has been shown to be an endogenous ligand of the apelin receptor (APJ) [29]. Expression of this receptor system has been described in central nervous system, and is several peripheral tissues, including endothelium. Furthermore, expression of apelin is found in cardiomyocytes and vascular cells; thus playing a key role in the regulation of vascular tone and cardiovascular function [20]. While apelin has been reported to be upregulated by insulin, it has also been found to be secreted by adipocytes, and has thus been considered as an adipokine, which has generated questions about its role in metabolic disorders [30].
Folistatin-Like 1
(FSTL-1) has an antagonistic effect on the transformation of transforming growth factor beta (TGF-β) family, a large group of proteins that includes activin, myostatin, and bone morphogenetic protein (BMP). FSTL-1 has been associated with an improvement in vascular function, counteracting endothelial dysfunction. It is also known, as osteonectin, to antagonize myostatin, and high levels of FSTL-1 have been found to be protective against cardiovascular disease [31].
Irisin
Irisin is highly expressed in skeletal muscle and adipose tissue, and is secreted during muscle contraction. Irisin is related to the increase in energy expenditure, due to its ability to transform white fat into brown by increase protein levels of uncoupling protein 1 (UPC1) [32,33]. An effect has also been found in the myocardium, where irisin protects against ischemia, cardiac damage, improves ventricular function after ischemia, increases coronary flow, decreases the area of infarction, decreases levels of caspase 3 and annexin V, and phosphorylates p38 MAPK and SUP-1 in the ischemic myocardium [34]. High plasma levels of irisin lead to a marked improvement of glucose tolerance and a decrease in body weight; these two results induce the change of white adipose tissue in brown, leading to an increase in energy expenditure and favoring the utilization of that brown fat as an energy source during exercise [35].
Myostatin
Myostatin is a member of the transforming growth factor-beta superfamily/bone morphogenetic protein (TGF-β/BMP) family member, growth factor/differentiation 8 (GDF-8). This gene was called myostatin because of its ability to inhibit differentiation and muscle growth; with regulatory capacity of metabolism and that its inhibition can significantly attenuate the progression of obesity and diabetes. Treatment with myostatin increased glucose uptake and glycolysis, and inhibited glycogen synthesis in skeletal muscle cells cultured in vitro through an AMP kinase-dependent mechanism [36,37]. Although at least some of these effects on metabolism can be attributed to the influence of myostatin on skeletal muscle growth and, therefore, on the total volume of metabolically active body mass, there is growing evidence that myostatin affects the growth and the metabolic state of other tissues, including adipose and liver. In addition, recent work has explored the role of myostatin in the mobilization of the substrate, the uptake and/or utilization of the muscle, independently of its effects on the body composition. Finally, the effects of myostatin and resistance exercise as well as the potential role of myostatin in the beneficial metabolic adaptations that occur in response to exercise have also begun to be delineated in greater detail [38].
Hepatokines
While among organokines, adipokines were the first to be described, knowledge of new hormonal mediators of insulin-sensitive tissues involved in adiposopathy has emerged, recognizing the existence of myokines, cardiokines, and hepatokines [6]. Hepatokines are proteins secreted predominantly by the liver, although they are also produced by placenta and tongue in minimal amounts with paracrine and endocrine action [36]. Some authors claim that hepatokines have an ambivalent role, since on one hand they promote insulin resistance and on the other, they favor metabolic variability of type 2 diabetes mellitus [37].
The first described hepatokine was Fetuin-A, which was isolated from bovine serum in 1944. In humans, fetuin-A is a 64 kDa glycoprotein encoded by the AHSG gene. Levels of fetuin-A are increased in metabolic syndrome, obesity, and type-2 diabetes mellitus by direct inhibition of phosphorylation of the signalling cascade and translocation of GLUT-4 receptors in the other target tissues of insulin [39]. A more recent study has described that genetically, the association between the 3q27 loci in which Fetuin-A is found- and its relationship with the metabolic syndrome and type 2 diabetes has been proved, confirming that the absence of the coding gene improved insulin signalling [38-40]. This hepatokine acts as an endogenous inhibitor of Toll receptor type 4 (TLR4), a receptor tyrosine kinase and inflammatory mediator, promoting the release of fatty acids, proinflammatory signalling, and insulin resistance [41,42]. Experimental evidence has detected over-expression of fetuin-A to be induced in rodents fed with obesogenic diet, identifying the association between increased levels of fetuin-A and non-alcoholic fatty liver disease [43].
Likewise, Haukeland et al. [44] reported an association of the expression of fetuin-A in liver with enzymes involved in carbohydratelipid metabolism. Additionally, they identified a relationship between fetuin-A and homeostasis model assessment of insulin resistance (HOMAIR) values with serum triglyceride levels [45]. This suggests a potential point of study where fetuin-A is present, as a possible trigger of fatty infiltration, even prior to the presentation of liver disease, and possible predictor of type-2 diabetes mellitus [46]. Already under the pressure of non-alcoholic liver disease, high concentrations of fetuin- A, compared with control groups, has been confirmed [47,48].
Another widely recognized and studied hepatokine is fibroblast growth factor 21 (FGF-21). Synthetized mainly in the liver, FGF-21 has been shown in mouse models to increase peripheral glucose uptake by favoring insulin sensitivity, to reduce hepatic and serum triglycerides, as well as glucose, achieving greater caloric expenditure and therefore weight loss [49]. In addition, FGF-21 has been shown to increase insulin sensitivity in peripheral muscle and liver tissue independently of the effect on weight and fat [50]. Altogether, these data suggest that future studies of substances with insulin-like activity that could contribute to counteract the obesogenic environment of insulin resistance should be carried out.
Hepatic insulin-sensitizing substance (HISS), a hepatokine that has been described by some authors as being key in the development of insulin resistance, indicating that, as the lack of insulin is the cause of diabetes type 1, the absence of HISS would be related to the progression to type 2 diabetes [51]. This hypothesis suggests that postprandial insulin action is altered up to 55% with the blockade of HISS, its effect therefore being dependent insulin. In the postprandial physiological state, HISS stimulates skeletal muscle glucose uptake and glycogen formation, as well as the redistribution of glycogen storage, Therefore, the lack of HISS leads to increased hepatic triglyceride synthesis secondary to the inability to store glycogen. HISS activity depends on the intrinsic insulin effect and the parasympathetic hepatic innervation of which, curiously, it is lacking in fasting conditions, resulting in HISS-dependent insulin resistance (HDIR) [51].
Insulin resistance to HISS is physiologically present during fasting, pregnancy, or trauma. Food intake activates this cascade, which decreases in fasting situations, decreasing the hepatic nervous stimulation resulting in safe release of HISS. The hormonal action of HISS is based on observational studies in which hepatic nerve signalling was simulated using acetylcholine and portal vein denervation in rodent (rat) and canine models, in which insulin action was restored [52]. However, that study indicated the failure to reverse insulin resistance with intravenous acetylcholine, indicating its action in hepatic receptors. On the contrary, an additional experiment performed in rodent (rat) and rabbit models showed the effect of intravenous hepatic nitric oxide with evidence of restoration of resistance to HISS [53]. Altogether, these data indicate a hormonal relationship between liver and muscle before the persistence of the mechanism of insulin despite the breakdown of the neural link and humoral action in peripheral tissues.
Adipokines
Adipose tissue is a very active tissue that secretes multiple proteins known as "adipokines", which are involved in insulin resistance, and therefore in cardiovascular complications [54].
However, not only adipokines are involved in this process, since at a cellular level infiltrating macrophages in the adipose tissue make obesity comparable to a chronic inflammation, thus evidencing a link between adipose cells and the immune system [55]. One of the main adipokines is leptin, a protein encoded by the OB gene on chromosome 7 that acts as a "lypostate" [56]. This means that when stored fat in adipocytes increases, leptin is released into the bloodstream and informs the hypothalamus that the body has enough reserves, which translates into inhibition of appetite. Adipose tissue and plasma leptin levels depend on the amount of energy stored as fat and the state of energy balance. Therefore, leptin levels are higher in obese individuals and increase with over-feeding, while lower leptin levels are found in thin individuals. Thus, it is inferred that fasting results in a reduction of circulating leptin [57]. A pro-inflammatory response has been observed in high leptin levels, which could explain the possible control of leptin on tumour necrosis factor (TNF), as well as the production and activation of macrophages [6,58]. Furthermore, additional studies show that leptin could trigger the synthesis of endothelin-1 and nitric oxide (NO) synthase, as well as the production of monocyte chemotactic protein (MCP-1), proliferation and migration of endothelial cells, platelet aggregation, and the accumulation of cholesterol in the macrophages that will result in hyperglycemia and in angiogenesis, to finally lead to arterial vessel damage [59-62]. On the other hand, leptin improves insulin sensitivity by activating the AMP-activated protein kinase (AMPK), which controls cellular concentrations of malonyl-CoA, thus inhibiting acetyl-CoA carboxylase (an enzyme involved in the transformation of malonyl- CoA) [63].
As a result, there is a decrease in intracellular malonyl-CoA and a decrease in lipogenesis associated with an increase in fatty acid betaoxidation. In fact, in generalized lipodystrophy, where adipose tissue is almost absent, administration of leptin improves insulin sensitivity [64].
However, despite this hypothesis, administration of leptin has little or no effect on insulin resistance. Another hypothesis postulated that the effect of insulin can be altered by the leptin signalling pathway, which activates suppressor of cytokine signaling-3 (SOCS-3), and could thus inhibit insulin signalling [65].
TNF-α is a pro-inflammatory cytokine produced by numerous cells, but mainly by macrophages and lymphocytes, and to a lesser extent by adipose tissue. Insulin sensitivity is mediated by abnormal phosphorylation of the insulin receptor substrate (IRS) by altering the beta cell and favoring gluconeogenesis. Although poorly expressed in human adipose tissue, TNF-α can accelerate atherosclerosis, mainly through the induction of the expression of the vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule (ICAM) in cells of vascular and endothelial smooth muscle, resulting in altered endothelium-dependent vasodilatation and in the promotion of endothelial cell apoptosis [66,67].
IL-6 it is produced by several cells and to a lesser extent by adipose tissue. The main function of IL-6 is the hepatic production of inflammatory proteins such as C-reactive protein (CRP), which would explain the pro-inflammatory process of central obesity in cardiovascular diseases [68]. Insulin resistance is mainly influenced by the alteration of signaling in hepatocytes by inhibiting autophosphorylation of insulin receptor (IRS) and phosphatidyl inositol-3 kinase (PI3K) through the activation of AMPK (protein kinase stimulated by AMP). This responds to constant energy changes affecting pancreatic beta cells, and the consequent alteration of the synthesis and secretion of insulin [69].
Resistin is a cysteine-rich protein that plays an important role in insulin resistance, hence its name. Resistin is a pro-inflammatory protein found at high levels of resistin in adipose tissue, which rise considerably once obesity is established [8]. However, there are contradictory positions regarding its effect [70,71]. In vitro studies have shown that insulin has a significant suppressive effect on resistin mRNA synthesis in adipocytes, and this occurs with insulin concentrations that lower than physiological levels [72].
Adiponectin, one of the most abundant adipokines, unlike the previously described proinflammatory adipokines, acts as an antiinflammatory substance. The simplest forms of human adiponectin include adiponectin trimer or low molecular weight (LMW), followed by the middle molecular weight (MMW), and high molecular weight (HMW) forms of the hexamers and multimers, respectively [73]. The latter is the most important because it is the one that most sensitizes insulin and, like leptin, it interacts with AMPK [74]. Adiponectin afects hepatic glucose synthesis by decreasing mRNA expression of two enzymes involved in gluconeogenesis; phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) [75]. It has an important anti-TNF effect, by which adiponectin exerts an important anti-atherosclerosis effect by acting directly on macrophages.
he term adiposopathy refers to a complex signaling network between organs, secondary to increase size of the fatty tissue with subsequent alteration in its auto-regulation and therefore inflammatory changes with subsequent clinical manifestation. While the term obesity is currently used in a generalized way, this concept is linked in clinical practice to the body mass index (BMI); in itself excluding the inherent organ damage of overweight or normal weight people with high body fat. he above could be explained by the fact that this entity is not addressed in a holistic view, which allows the location of fatty tissue (deleterious) or skeletal muscle (beneficial) to be discriminated. To be able to discriminate between the deleterious and beneficial events associated with adiposopathy, we postulate the CLARos physiopathological model, which brings together very active organs in charge of maintaining an adequate balance between glucose and insulin (the triumvirate).
In this way, the liver appears as organ associated with in the progression of this multifactorial pathology, with a high cardiometabolic impact, with an endocrine network that seeks the protective homeostasis of its pieces, which is highly sensitive to metabolic disruption.
In adiposopathy, hepatokines and adipokines are elevated, and establish a close relationship between liver and adipose tissue. hese cytokines exert superiority over the myokines that would have in its majority a protective function, and therefore reduce its functionality and production.
This does not have a direct relationship according to weight, but does have association with quantity of fatty tissue and insulin resistance, resulting in a pro-inflammatory environment, known as “dysmetabolism". his inflammatory microenvironment without clinical manifestations is reflected by high fatty component, where the endocrine regulation that maintains the homeostasis of glucose is preserved.
Therefore, the CLARos model seeks a balance between the compromised organs. The ideal point is achieved when each component of the triumvirate - myokines, hepatokines, and adipokines – is able to provide adequate information about the proper balance of functionality of its cytokines related to the normal functioning of insulin, without altering the substances of the signalling pathways. Thus, it strongly suggests that the intervention for this pathology should not only be pharmacological but, on the contrary, it requires multiple preventive and palliative intervention strategies that favor the recovery of high levels of myokines, decreasing pro-inflammatory adipokines (Figure 2) [77].
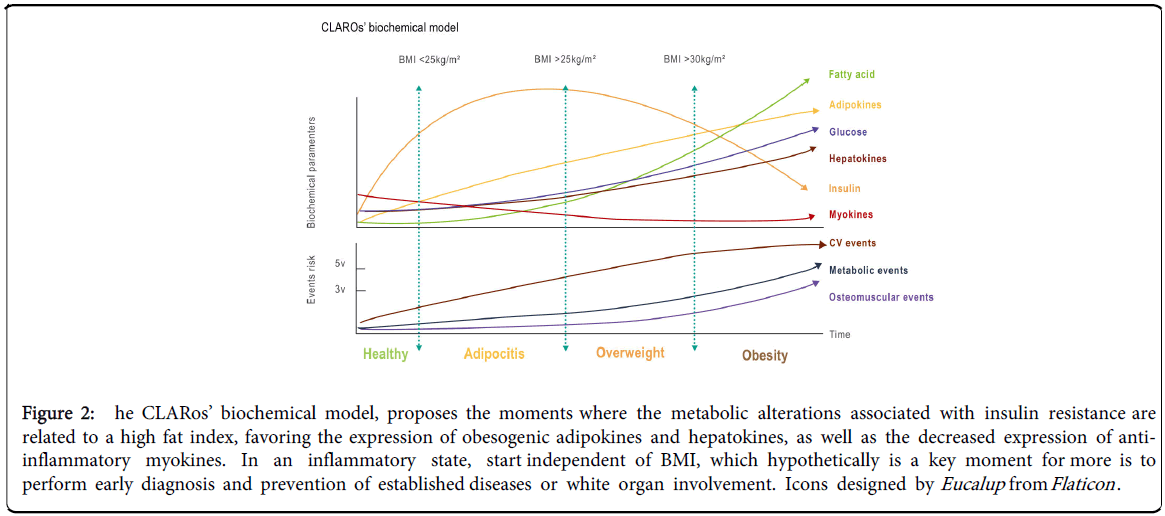
Figure 2: he CLARos’ biochemical model, proposes the moments where the metabolic alterations associated with insulin resistance are related to a high fat index, favoring the expression of obesogenic adipokines and hepatokines, as well as the decreased expression of antiinflammatory myokines. In an inflammatory state, start independent of BMI, which hypothetically is a key moment for more is to perform early diagnosis and prevention of established diseases or white organ involvement. Icons designed by Eucalup from Flaticon .
Obesity has been generally considered as a disease in which the energy balance (consumption and expenditure) is altered, which, although of great importance, does not take into account the changes in different organs associated with the increased excess of energy reserves. hese alterations lead to the adaptation of the different affected organs, which between them form an endocrine network that tends to perpetuate itself pathologically.
Understanding that pro-inflammatory adipokines are increased in macrophage-infiltrated adipose tissue and myokines and hepatokines are altered along with ectopic deposition of fat on these tissues and organs, the concept of obesity shifts away from the monocular and traditional approach of BMI as the only tool used to determine risk or diagnosis of a chronic and silent disease.
In this article we propose a model that may serve as a basis to generate an integral clinical vision, which allows performing a clinical approach in which an incipient damage can be detected by assessing the increase in adipose tissue, besides being able to identify possible markers or detection rates in early stages of adiposopathy.
However, future studies are required to evaluate the interaction of related hormonal routes in the inflammatory states secondary to the increase of fatty tissue in order to achieve preventive approaches in patients at risk or of mild inflammation secondary to adiposity, and ultimately positively impact prevention, diagnosis, and treatment of this disease in early stages.