Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Research Article - (2015) Volume 3, Issue 1
The classifications of Myeloproliferative Disorders (MPD) by the Polycythemia Vera Study Group (PVSG) and World Health Organization (WHO) used crude criteria for the diagnoses of Essential Thrombocythemia (ET), Polycythemia Vera (PV) and Primary Myelo Fibrosis (PMF). The PVSG and the 2007 WHO criteria for the diagnosis of ET and PVoverlook the very early prefibrotic stages of MPN. The 2008 European Clinical, Molecular and Pathological (2008 ECMP) criteria are sensitive for the detection of early stages of JAK2V617F trilinear Myelo Proliferative Neoplasms (MPN) and could delineate three stages JAK2V617F mutated ET: normocellular ET; ET with features of early PV (prodromal PV); and ET with Hypercellular Megakaryocytic Granulocytic Myeloproliferation (EMGM). The 2008 ECMP classification distinguishes six clinical PV stages that have important prognostic and therapeutic implications. Spontaneous EEC, low serum erythropoietin (EPO) levels and JAK2 mutations are highly specific for ET with PV features (prodromal PV), masked PV and classical PV. JAK2 wild type ET and MF have no blood and bone marrow features of PV.
The detection and quantitation of JAK2V617F mutation allele burden play a key-role in the diagnostic work-up and staging of ET, PV and MF patients. The JAK2V617F mutation allele burden in heterozygous mutated ET and in combined heterozygous-homozygous or homozygous mutated PV and EMGM is of major clinical and prognostic significance. Pre-treatment bone marrow histopathology is of huge importance to document and stage the broad spectrum JAK2 mutated and JAK wild type MPN. JAK2 wild type ET carrying the MPL515 mutation is a separate and distinct MPN entity of ET and MF without features of PV at diagnosis and during follow-up. JAK2 wild type hypercellular ET associated with Primary Megakaryocytic Granulocytic Myeloproliferation (PMGM) is the third MPN entity of elusive etiology. Myelofibrosis (MF) is not a primary MPN disease entity because Reticulin Fibrosis (RF) and Reticulin/Collagen Fibrosis (RCF) are a secondary response of polyclonal fibroblasts to cytokines released from the clonal granulocytic and megakaryocytic proliferative cells.
Keywords: Myeloproliferative neoplasm; Essential thrombocythemia; Polycythemia vera; Myelofibrosis; JAK2V617F mutation; MPL515 mutation; Bone marrow pathology; World Health Organization
In the 19th century Chronic Myeloid Leukemia (CML) and Polycythemia Vera (PV) have been described as primary distinct disease entities [1-3]. In 1951 Dameshek lumped dissimilar diseases of polycythemia vera, erythroleukemia, idiopathic and agnogenic myeloid metaplasia, megakaryocytic leukemia and proposed an unifying theory that all these variable manifestations represent one myeloproliferative activity of bone marrow cells due to one hypothetical stimulus (Figure 1) [3]. Lumping erythroleukemia with PV, and putting together Chronic granulocytic or Myeloid Leukemia (CML) with PV appeared to be without scientific foundation (Figure 1). In 1960 Nowell and Hungerford described the presence of a minute chromosome in leukemic cells called Philadelphia (Ph) chromosome after the city of discovery as a diagnostic clue CML [4]. Using banding techniques Janet Rowley (1973) discovered that the Ph chromosome originated from a translocation between the chromosomes 9 and 22, t(9;22)(q34;q11) [5]. Three Dutch investigators discovered that a hybrid gene is generated by the translocation consisting of the BCR gene on chromosome 22 and the ABL oncogene originating from chromosome 9 [6-8], which results in a BCR/ABL fusion gene with high tyrosine kinase activity and CML-transformation capacity [9,10]. Ninety-five percent of all CML patients are Ph+; 90% are Ph+/BCR/ABL+, 5% are Ph-/BCR/ ABL+, and 5% are Ph-/BCR/ABL-, the latter group usually diagnosed as atypical CML, juvenile CML, chronic neutrophilic leukemia or chronic myelomonocytic leukemia [11].
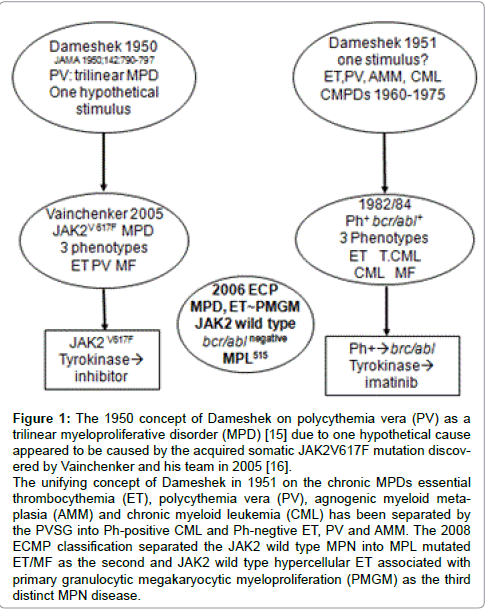
Figure 1: The 1950 concept of Dameshek on polycythemia vera (PV) as a trilinear myeloproliferative disorder (MPD) [15] due to one hypothetical cause appeared to be caused by the acquired somatic JAK2V617F mutation discovered by Vainchenker and his team in 2005 [16]. The unifying concept of Dameshek in 1951 on the chronic MPDs essential thrombocythemia (ET), polycythemia vera (PV), agnogenic myeloid metaplasia (AMM) and chronic myeloid leukemia (CML) has been separated by the PVSG into Ph-positive CML and Ph-negtive ET, PV and AMM. The 2008 ECMP classification separated the JAK2 wild type MPN into MPL mutated ET/MF as the second and JAK2 wild type hypercellular ET associated with primary granulocytic megakaryocytic myeloproliferation (PMGM) as the third distinct MPN disease.
The PVSG used in 1975 the Ph1-chromosome to separate the Ph1- negative ET, PV and AMM from the Ph1-positive ET and Chronic Myeloid Leukemia (CML) with various degrees of thrombocythemia and myelofibrosis [12,13]. According to strict morphological, biochemical, cytogenetic and molecular criteria including the Ph+ chromosome and bcr/abl fusion gene and protein, CML is a malignant disease with an obligate transition into acute leukemia, whereas Essential Thrombocythemia (ET), Polycythemia Vera (PV) and Agnogenic Myeloid Metaplasia (AMM) or Primary Megakaryocytic Granulocytic Myeloproliferation (PMGM) form the Ph-chromosome and BRC/ABL negative chronic Myelo Proliferative Disorder (MPD) featured by a benign proliferation of the three hematopoietic cell lines [14].
Regarding etiology of PV, Dameshek proposed two highly speculative possibilities: first, the presence of excessive bone marrow stimulation by an unknown factor or factors, and second, a lack or a diminution in the normal inhibitory factor or factors [15]. The discovery of the JAK2V617F mutation in 2005 by Vainchenker confirmed the hypothesis of Dameshek that PV indeed appeared to be a trilinear MPD by the disvery and demonstration that loss of inhibitory activity of the JH2 pseudokinase part on the JH1 kinase part of JAK2, leading to enhanced activity of the normal JH1 kinase activity of JAK2 as the cause of trilinear MPNs ET, PV and MF Figures 1 and 2) [16,17] The sequential stages of heterozygous and homozygous JAK2V617 mutations (Figures 1 and 2) render the TPO and EPO receptors constitutively activated and hypersensitive to hematopoietic growth factors Thrombopoietin (TPO), Erythropoietin (EPO) and Granulocyte Colony Stimulating Factor (GCSF), resulting in trilinear MPN (ET, PV and MF [16,17].
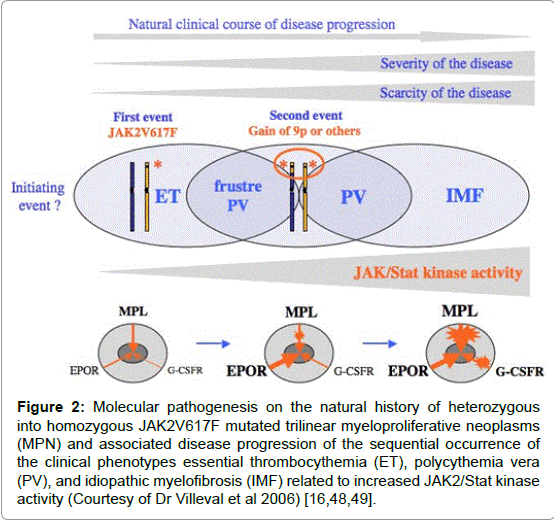
Figure 2: Molecular pathogenesis on the natural history of heterozygous into homozygous JAK2V617F mutated trilinear myeloproliferative neoplasms (MPN) and associated disease progression of the sequential occurrence of the clinical phenotypes essential thrombocythemia (ET), polycythemia vera (PV), and idiopathic myelofibrosis (IMF) related to increased JAK2/Stat kinase activity (Courtesy of Dr Villeval et al 2006) [16,48,49].
The morphological distinction between Ph+ and BCR/ABL+ ET and thrombocythemia associated with Ph+ and BCR/ABL+ CML versus the Ph-negative thrombocythemia in various MPDs is primarily based upon conspicuous differences in the form and size of megakaryocytes in bone marrow smears and sections of bone marrow biopsy [14]. This difference in bone marrow smears observed by Michiels et al in 1987 is reproducible in bone marrow biopsies by pathologists to distinguish between small mononucleated megakaryocytes in Ph+ diseases versus clustered large megakaryocytes with hyperlobulated nuclei in Phnegative MPDs [14,18-26]. Georgii et al defined in 1990 [22] the Hannover Bone Marrow classification of the MPDs, which had the great advantage to pick up the early stages of prefibrotic MPD ET,PV and PMGM, which are overlooked by the PVSG criteria. Thiele introduced in 2001 bone marrow features on top of PVSG criteria and defined the World Health Organization (2001 WHO MPD) classification [25,26]. The PVSG [12,13], the 2001 WHO [26] and the 2007 WHO [27] criteria are rather crude to adequately classify the MPDs as ET, PV and CIMF and overlook the very early prefibrotic stages of MPD by definition [19-23]. In the present manuscriptF we could integrate the PVSG and the 2001 and 2007 WHO criteria into the 2008 ECMP classification (Tables 1-3) by including bone marrow pathology and the use of specific laboratory features and molecular markers for diagnostic differentiation of each of the latent (masked), early and overt MPDs or MPNs.
| Clinical and molecular criteria | Bone marrow pathology (P) criteria (WHO) |
| JAK2V617F ET | Normocellular ET |
| 1.Platelet count of >350 × 109/l and the presence of large platelets in a blood smear 2.Presence of JAK2-V617F mutation 3.Normal erythrocytes <5.8× 1012/L males, <6× 1012/L females 4.Normal haemoglobin(Hb) and hematocrit (ht) |
Predominant proliferation of enlargedmature megakaryocytes with hyperlobulated nuclei and maturecytoplasm, lacking conspicuous morphological abnormalities. No increase, proliferation or immaturity of granulopoiesis orerythropoiesis. Reticuline fibrosis (RF) 0 or 1 |
| JAK2V617F prodromal PV | ET with bone marrow features of PV |
| 1.Platelet count of ≥350× 109/l andnormal ht male <0.51, female <0.48, normal erythrocyte <5.8× 1012/L males, <6× 1012/L females is mandatory. 2.Presence of JAK2-V617F mutation 3.Low serum EPO level and/or increased LAP score 4.Spontaneous EEC. |
Increased cellularity with due to icreasederytropoiesis or trilineagemyeloproliferation (i.e. panmyelosis). Proliferation and clustering of small to giant (pleomorphic) megakaryocytes. Absence bone marrow features consistent with congenital polycythemia and secondary erythrocytosis. RF 0 or 1 |
| JAK2V617F hypercellular ET | ET.MGM |
| 1.Platelet count of≥350× 109/l, 2.No signs of leuko-erythroblastosis 3.Slight or moderate splenomegalyon ultrasound 4.Presence of JAK2-V617Fmutation 4.No preceding or alliedCML, PV, RARS-T or MDS. ET.MGM clinical staging: Early stage: No anemia with hb and ht in the normal low normal range: hb>13g/dl: early clinical stage Intermdiate:Hb<13 to >12 g/dL, LDH N or ↑, no leukoerythroblastosis Advanced:Hb<10 g/dL, LDH↑↑, CD34+ , leukoerythroblastosis, tear drop |
Hypercellular ET due to chronic megakaryocytic and granulocytic myeloproliferation (EMGM) and normal or reduced erythroid precursors. Loose to dense clustering of more pleiomorphic megakaryocytes with hyperploid or clumpsy nuclei (not or some cloud-like). RF grading PVSG, MF Georgii and Thiele (Table 3) Prefibrotic: RF- 0/1, MF-0, no/minor splenomegaly Bone marrow staging: Early fibrotic ET:RF 2, MF 1, splenomegaly no/minor Fibrotic ET: RF3, RCF or MF2, overt splenomegaly Post-ET MF: RF3/4, or MF-2/3 |
Table 1: The 2008 WHO Clinical Molecular and Pathobiological (WHO-CMP) criteria for the diagnosis JAK2V617F mutated essential thrombocythemia (ET).
| Clinical criteria JAK2 mutated PV A1 Erythrocyte count above 6 × 1012/L, hemoglobin .18.5 g/dL male and >16.5 g/dL females. Raised red cell mass (RCM optional) male .36 ml/kg, female >32 ml/kg (PVSG, WHO) A2 Persistent increase of platelet count grade I 400-1500, grade II >1500× 109/L A3 Splenomegaly on ultrasound or CT (>12 cm) or splenomegaly on palpation A4 Granulocytes >10× 109/L or leukocytes >12× 109/L and raised LAP score >100 in the absence of fever and no increase of ESR A5 Absence of any cause of primary or secondary erythrocytosis A6 Low plasma or serum EPO level Clinical criteria MPL515 mutated ET A1 Persistent increase of platelet count grade 1 400-1500, grade II >1500× 109/L A2 Normal spleen or only minor splenomegaly on echogram A3 Normal LAP score, normal ESR and increased MPV A4 Spontaneous megakaryocyte colony formation (CFU-Meg) A5 No signs or cause of reactive thrombocytosis A6 No preceding or allied other subtype of MPN, PV, MDS or CML A7 Absence of Philadelphia chromosome Staging according to no, mild or severe anemia Clinical criteria JAK2 wild type ET andPMGM A1 No preceding or allied other subtype of MPN, PV, CML or MDS, JAK2 and MPL wildtype Early clinical stage: no anemia Normal hemoglobin, or anemia grade I: hemoglobin >12 g/dL, slight or moderate splenomegaly on palpation or >11 cm on ultrasound or CT. Thrombocythemia>400× 109/L Intermediate clinical stage; mild anemia Anemia grade II, hemoglobin >10 g/dL, definitive leuko-erythroblastic blood picture and/or tear-drop erythrocytes. Splenomegaly on palpation, no adverse signs Advance clinical stage: severe anemia Anemia grade III, hemoglobin<10 g/dL, significant splenomegaly and one or more adverse signs |
Pathological criteria PV B1 Increased cellularity due to increased erythropoiesis or due to trilinearmyeloproliferation of megakaryopoiesis, erythropoiesis and granulopoiesis (e.g. panmyelosis). Proliferation of small medidium sized and large (pleomorphic) megakaryocytes.Absenceofstainable iron, No or slight increase of reticulin fibers. B2 Sponataneouserythroid colony (EEC) formation A1 + B1 and none of the others is idiopathic erythrocythemia: IE A2 + B1 and none of the others is ET with features of PV (prodromal PV) A3 and B1 and none of the other is primary MPD or latent PV A1 + B1 plus one of A2 to A6 or B2 is overt classical PV Pathological criteria MPL515 mutated ET B1 Predominant proliferation of enlarged to giant megakaryocytes wit hyperlobulatedstaghorn-like nuclei and mature cytoplasm, lacking conspicious cytological abnormalities B2 No proliferation or immaturity of granulopoisis or erythropoiesis B3 No or only borderline increase in reticulin fibers The combination of A1 and B1 + B2 establish ‘true’ ET. Any other criterion confirms ET. LAP=leukocyte alkaline phosphatase; ESR=erythrocyte sedimentation rate; MPV=mean platelet volume; MPN=myeloproliferative neoplasm; PV=polycythemia vera; MDS=myelodysplastic syndrome; CML=chronic myeloid leukemia; Staging myelofibrosis (MF) according to MF grading Pathological criteria JAK2 wild type PMGM B1 Megakaryocytic and granulocytic myeloproliferation (MGM) and relative or absolute reduction of erythropoiesis (erythroid precursors. Abnormal clustering and increase of atypical immature medium-sized large to giant megakaryocyte containing (Cloud-like) hypolobulatednucle and definitive maturation defects Staging of myelofibrosis: MF in PV and PMGM MF 0 no reticulin fibrosis RF 0/1 MF 1 slight reticulin fibrosis RF 2 MF 2 marked increase RF grade 3 and slight to moderate collagen fibrosis MF 3 advanced collagen fibrosis-osteosclerosis (endophytic bone formation) |
Table 2: The 2008 European Clinical and Pathological (2008 ECMP) criteria for the diagnosis of JAK2 mutated polycythemia vera (PV), MPL515 mutated ET [3-5] and JAK2/
MPL wild type hypercellular ET associated with primary megakaryocytic granulocytic myeloproliferation (PMGM).
| USA Subjective |
UK Subjective |
Grading of myelofibrosis (MF) Descriptive: silver impregnation, Masson stain |
MF |
| RF 1 | RF 1+ | Scattered linear fine fibers with no intersections (cross-overs) and rare course reticulinfibers |
MF 0 Prefibrotic |
| RF 2 No Masson stain |
RF 2+ RF 3+ No Masson stain |
Loose network of reticulin with intersections around megakaryocytes and in perivascular areas: silver impregnation no collagenisation: Masson stain |
MF 1 Early reticulin fibrosis: RF |
| RF 3 No Masson stain |
RF 4+ Dry tap |
Diffuse and dense increase in reticulin with extensive intersections, occasionally only focal bundles of collagen and/or focal osteosclerosis | MF 2 Fibrotic |
| RF 4 No Massaon Stain |
Dry tap | Diffuse and dense increased in reticulin with extensive interactions with coarse bundles of collagen, often associated with significant osteosclerosis | MF 3 Sclerotic |
Table 3: Grading of reticulin fibrosis (RF) according to Ellis [34], and European grading of myelofibrosis (MF) according Thiele et al. [75] in bone marrow biopsies of patients with a chronic myeloproliferative neoplasms (MPN).
With the improvement of bone marrow biopsy and tissue processing in the 1980s and 1990s, Georgii and Thiele defined the pathological features of ET, PV and CIMF or AMM on bone marrow histopathological morphology [18-24]. Georgii regarded Myelofibrosis (MF) as a reactive feature secondary to progressive disease [18] seen in AMM, PV and CML [22-24]. Georgii reasoned that the terms Agnogenic Myloid Metaplasia (AMM) or CIMF lack accuracy since they are applied to both the pre-fibrotic hyper cellular and advanced fibrotic stages [22]. As Myelofibrosis (MF) is not a disease but a secondary complication of MPD he used the term MF for grading of the MPD disease burden based on the degree of Reticulin Fibrosis (RF) and reticulin-collagen fibrosis (Tables 1-3). Accordingly, Georgii et al replaced the terms AMM and CIMF by chronic or primary megakaryocytic granulocytic myeloproliferation (CMGM or PMGM). The Hannover Bone Marrow classification have defined ET by persistent increase of platelets in excess of 400 x109/l without the Ph+ chromosome together with monolinear proliferation of mature enlarged megakaryocytes in the bone marrow with normal cellularity, normal erythropoiesis and normal granulopoiesis [22-24]. The Hannover Bone Marrow classification have defined PV as a trilineage proliferation of megakaryopoiesis, erythropoiesis and granulopoiesis in which the erythropoiesis was most prominent together with variable degrees of increased platelets, erythrocytes and granulocytes in the peripheral blood in the absence of the Ph+ chromosome [22-24]. The diagnosis of prefibrotic CMGM/PMGM [18-20] has been labelled as chronic idiopathic myelofibrosis (CIMF) in the 2001 WHO classifications [26]. The diagnosis of CMGM/PMGM according to the 1996 Hannover [22-30] and the 2008 ECMP classification respectively are based on three specific bone marrow histology criteria: 1) the presence of large dysmorphic megakaryocytes with immature cytoplasm and immature cloud-like nuclei not seen in ET and PV, 2) increased granulopoiesis but never disturbed in maturation and 3) no features of PV with relatively decreased erythropoiesis.
The PVSG failed to use BMB and introduced 3 major and 4 minor clinical criteria as inclusion criteria the diagnosis of PV in the PVSG- 01 study of which increased RCM was mandatory [12,13]. Increased RCM in PV patients corresponded to hematocrit values between 0.48 and 0.76 in all, platelet count above 400 x109/L in two-third and palpable spleen in two-third of about 400 PV patients in the PVSG-01 study [13]. The PVSG and WHO classifications excluded ECP defined Idiopathic Erythrocythemia (IE) by definition [31,32]. IE is featured by increased hemoglobin, haematocrit, erythrocytes and increased red cell mass but normal leukocytes, thrombocytes and spleen size on palpation [31,32]. Minor B criteria did appear in untreated IE patients during follow up and was associated with a high incidence of major or lethal cerebrovascular thrombotic disease 31. This category of IE or the earliest erythrocythemic stage of PV comprises about 10 to 20% of the PV cases at time of presentation [31,32]. In the 1970s tools available to separate the Idiopathic Erythrocytoses into myeloproliferative PV (IE) versus primary or secondary erythrocytosis by including pretreatment bone marrow biopsy [33-35] and EEC 36,37 were overlooked by the PVSG. Clinicians and pathologists should be aware that pretreatment bone marrow biopsy specimens in 191 PV patients of the PVSG-01 study [34,35] with increased RCM showed a normal bone marrow cellularity with no increase of clustered large megakaryocytes (idiopathic erythrocytosis) in about 7.5%, slight to moderate increased bone marrow cellularity (60 to 80%) and increase of clustered large megakaryocytes in two-thirds (consistent with early erythrocythemic, thrombocythemic stage PV), and pronounced trilineage hypercellularity (80 to 100%) of the bone marrow in one-third (consistent with classic PV with trilinear “panmyelosis” as described by the PVSG exactly as defined at the bone marrow level by Georgii et al [22-24], Michiels & Thiele [36-40] and by Thiele et al [41-43].
Between 2000 and 2007 it turned out that various degrees of characteristic PV bone marrow histology features (irrespective of RCM measurements) are seen in the four different stages of newly diagnosed patients with the JAK2 muated MPN (Tables 1, 2 and 4) [44- 47]. First, early thrombocythemic PV mimicking ET with a hematocrit in the upper limit of normal (< 0.50) but increased platelet count (> 400 x109/l) without or with slight splenomegaly (stage 0 PV); Second, “idiopathic erythrocytosis” with increased RCM, high hematocrit, low serum EPO, but normal platelet count and spleen size (stage 1 PV, Table 4); Third, classic PV with increased RCM, high hematocrit and one or more B criteria (overt stage 2 and 3 PV, Table 4); Fourth, unclassifiable or masked MPD with splenomegaly, normal hemoglobin and hematocrit, normal or slightly elevated platelet count (Iapparent PV=IPV, Table 4). Thiele et al confirmed that characteristic PV bone marrow histopathological features are seen in classic PV and in latent PV or early thrombocythemic PV mimicking ET [42] .
| PV: WHO-ECMP stage | 0 | 1 | 2 | 3 | 4 | 5 | 6 |
| WHO-ECMP Clinical Diagnosis |
Prodromal PV |
Erythrocy-themic PV | Early PV | Manifest PV Classical PV |
PV early MF Masked PV |
Inapparent PV |
SpentPV Post-PV MF |
| LAP-score | ↑ | ↑ | ↑ | ↑ | ↑/↑↑ | ↑ | variable |
| EEC | + | + | + | + | + | + | + |
| Serum EPO | N/↓ | N/↓ | ↓ | ↓ | ↓ | ↓ | variable |
| Erythrocytes x1012/l | >5.8 | <5.8 | >5.8 | >5.8 | >5.8 | Normal <5.5 | Decreased |
| Leukocytes x109/l | <12 | <12 | <or >12 | < or->15 | >15 | N or ↑ | >20 |
| Platelets x109/l | >400 | ,400 | < or >400 | >400 | < or >1000 | N low or ↑ | variable |
| WHO-ECMP bone marrow | Early PV | Early PV | Early PV | Trilinear PV | Trilinear PV | Prilinear PV | Myelofibrosis |
| Bone marrow cellularity (%) | 50-80 | 50-80 | 60-100 | 80-100 | 80-100 | 60-100 | Decreased |
| Grading reticulin fibrosis: RF | RF 0-1 | RF 0-1 | RF 0-1 | RF 0/1, | RCF1/2/3 | RCF 1/2/3 | RCF 3/ 4 |
| Grading myelofibrosis: MF57 | MF 0 | MF 0 | MF 0 | MF 0 | MF 0/1 | MF 0/2 | MF 2/3 |
| Splenomegaly on palpation | No/+ | No | No/+ | + | ++/+++ | ++/+++ | /large |
| Spleen size, echogram cm | <12-15 | <13 | 12-15 | 12-16 | 18->20 | 16 >20 | >20 |
| Spleen size on palpation cm | 0-3 | NP | 0-3 | 4-6 | >6 | >6 | >8 |
| JAK2V617F in Granulocytes% | Low | Low | Moderate <50 | High >50 | High >50 | Mod/High | High >50 |
| JAK2V617F in BFU-e (exon 12) | +(++) | +(++) | +(++) | ++ | ++ | + | ++ |
| Risk stratification → Therapeutic implications Anno 2014 |
Low risk | Low risk | Low risk | Intermediate risk PV | High risk PVearly MF |
Wait/See IFN JAK2 |
Post-PV MF Spent phase PV |
| First line Aspirin/Phlebotomy Second line IFN versus Hydroxyurea (HU) Third line JAK2 inhibitor |
Aspirin Phlebotomy |
Aspirin Phlebotomy |
Phlebotomy Aspirin Low dose IFN → responsive |
Phlebotomy* Aspirin IFN →resistant → HU |
If IFN resistant → HU or JAK2 inhibitor |
IF IFN Resistent JAK2 inhibitor |
JAK2 Inhibitor → Bone marrow transplant |
Table 4. Staging of JAK2V617F positive prodromal PV, erythrocythemic PV, classical PV, early MF, inapparent PV, spent phase PV and post-PV myelofibrosis (MF) according to WHO-ECMP criteria related to therapy anno 2008 (Black) and 2014 (Red) [78-80].
The 2007 WHO revision of the PVSG and 2001 WHO classifications decided to lower the platelet counts from 600 to around 450x109/l for the diagnosis of ET [27]. The 2007 WHO changed the term MPD into myeloproliferative neoplasia (MPN) and only defined the minimal criteria for ET, PV and Primary Myelofibrosis (PMF). We could improve the 2007 WHO revision of the MPNs the introduction of the 2006 → 2008 ECMP criteria for diagnoses of three distinct MPNs: JAK2V617F mutated ET and PV with various degrees of MF; JAK2 wild type ET and MF carrying the MPL515 mutation; and JAK2/ MPL wild type PMGM with features of hypercellular ET and various degrees of MF and splenomegaly (Tables 1-3) [27-32]. As compared to the 2008 ECMP classification, the 2007 WHO criteria by Tefferi et al are crude and not specific enough for three reasons. First, for ET they only include normocellular ‘true’ ET, but the diagnoses of early thrombocythemic PV (hemoglobin <18.5 for men and <16.5 for women) and ET associated with prefibrotic PV or MGM bone marrow (MF-0) with no leukoerythroblastosis, anemia or myelofibrosis (MF-0) remain unclassifiable. Second, the 2007 WHO criteria for PV arbitrarily exclude the early idiopathic stage of PV and overlooked masked PV just by the main crude inclusion criterion of a high hemoglobin level disregarding the importance increase of leukocytes, platelets and spleen size as typical features of masked trilinear PV. Simple tests like blood cell counts including platelets, leukocytes, hematocrit and erythrocytes above 6x1012/L40, and spleen size on echogram are not taken into account to distinguish the early thrombocythemic and erythrocythemic stages of PV from the classical overt trilinear polycythemic stage of classic PV as documented by bone marrow biopsy showing typical erythroid, granulocytic and megakaryocytic myeloproliferation [46,47]. Third, the 2007 WHO criteria defined Primary Myelofibrosis (PMF) as an endstage MPN disease complicated by anemia and splenomegaly, but the third prefibrotic entity of so-called prefibrotic PMGM (MF- 0) without leukoerythroblastosis or anemia remained unclassifiable. These shortcomings of the 2007 WHO diagnostic criteria for MPN by Tefferi et al [27] will hamper to prospectively evaluate the natural history, and therapeutic implications by objective staging of ET and PV MPN disease burden related to therapy. To overcome the shortcomings of the PVSG and WHO classifications of the MPDs and MPNs respectively, we here update and extend the ECMP criteria for the diagnosis, classification and staging of true ET, PV and PMGM (Tables 1-4).
The one cause hypothesis of trilinear PV proposed by Dameshek in 1950 [15] has been confirmed by Vainchenker in 2005 by the discovery of the JAK2V617F mutation as the driver cause of ET, PV, masked PV and MF (Figures 1 and 2) [16,17]. Detection of JAK2V617F has become the first intention diagnostic test to differentiate between PV and myeloproliferative Idiopathic Erythrocythemia (IE) from erythrocytosis with a sensitity of 95% and specificity of 100%. The discovery of the JAK2 V617F mutation by James et al [16] was immediately appreciated as an evolutionary event, and rapidly confirmed by several investigators (reviewed by Michiels et al 2006) [45]. JAK2 plays an essential role in cytokine-induced signalling from receptors to the nucleus by several hematopoietic cytokines including Erythropoietin (EPO), Thrombopoietin (TPO), and granulocyte colony stimulating factor (G-CSF) [48-50]. The JAK2V617F mutation renders the receptors on mutated hematopoietic progenitor cells hypersensitive to these cytokines, thereby resulting in growth advantage of the mutated aver the non-mutated trilinear hematopopoietic cells present in the bone marrow. The JAK2V617F mutation is detectable in CD34+ hematopoietic bone marrow cells, erythroblasts, in cells of spontaneous EEC, blood platelets and granulocytes (Table 5). Applying allele-specific Polymerase Chain Reaction (PCR) analysis in PVSG-defined MPD patients, a high frequency of the JAK2V617F mutation of 95% (92-97%) is described in PV, and a lower frequency of 53% (49-57%) in ET and 52% (44-55%) in MF [50,51]. Only 3 to 4% of ET, 24 to 27% of PV and 6 to 18% of MF patients are homozygous for the JAK2V617F mutation [50,51].
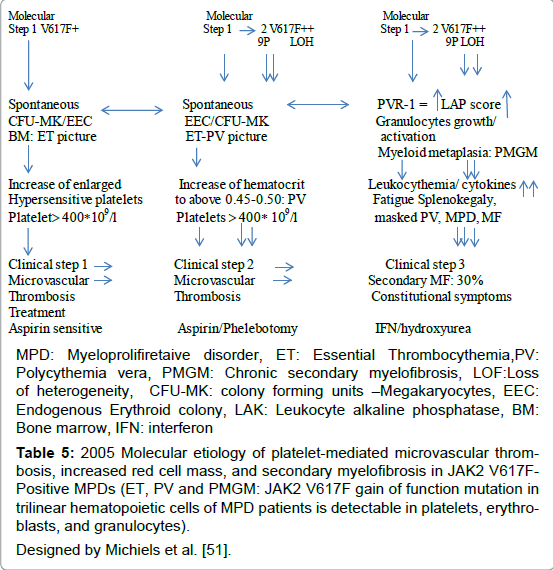
Table 5: 2005 Molecular etiology of platelet-mediated microvascular thrombosis, increased red cell mass, and secondary myelofibrosis in JAK2 V617FPositive MPDs (ET, PV and PMGM: JAK2 V617F gain of function mutation in trilinear hematopoietic cells of MPD patients is detectable in platelets, erythroblasts, and granulocytes). Designed by Michiels et al. [51].
Based on animal studies and different mutation states of JAK2V617F in MPN patients, two hypotheses have been proposed to explain why three different phenotypes of MPN are caused by the same JAK2V617Fmutation: the “dosage” hypothesis and the “additional events” hypothesis (Figure 3) [48,49]. According to the “dosage” hypothesis the level and duration of JAK2V617F directly contribute to the phenotypic diversity of trilinear MPNs (Table 5). This hypothesis is based on different densities of TPO receptors (TPOR or MPL)) and EPO receptors (EPOR) on hematopoietic progenitor cells and on differences of response of TPOR and EPOR to various levels of JAK2V617F activity [48,49]. TPOR/MPL is expressed at high levels in megakaryocytic cells where it controls physiological TPO levels. It is possible that activation of a few TPO receptors by low levels of JAK2V617F (heterozygous) is sufficient to send a signal to megakaryocytic cells [51]. A slight increase in numbers of mutated large (giant) megakaryocytes and platelets (about 50 to 100 x109/l mutated platelets) might be sufficient to produce platelet-mediated microvascular circulation disturbances (Table 5). Conversely, EPOR is expressed at low levels on hematopoietic progenitor cells and therefore high levels of JAK2V617F may be required to activate EPOR and generate a PV-like phenotype [48-52]. Sustained high levels of JAK2V617F during long-term follow-up subsequently may lead to a high level activation of activation of EPOR and GCSF receptor (GCSFR) leading to extramedullary hematopoiesis (splenomegaly) and cytokine mediated secondary myelofibrosis (Figure 3 and Table 5). The percentage of JAK2V617F positivity and progression from heterozygous to homozygous is strongly correlated with the ability to form spontaneous EEC formation and with progressive post-PV myelofibrosis. Scott et al. showed that BFU-e colonies are heterozygous in ET patients and did not contain a subpopulation of JAK2V617F homozygous cells (Figure 4) [52]. The BFU-e colonies are already homozygous for the JAK2V617F mutation in PV patients with a heterozygous pattern of JAK2V617F in their peripheral blood granulocytes (Figure 4) [52]. French investigators studied a large group of JAK2V617F positive PV (N=159, 36% homozygous) and ET (N=147, 4% homozygous), and genotyped BFU-E colonies in in 20 PV and 6 ET patients [53]. They showed that JAK2V617F positive ET patients usually harbour heterozygous BFU-E clones, some PV patients have a purely heterozygous profile, and most PV patients have a mixture of heterozygous and homozygous BFU-E clones [53]. Mutated erythroid progenitors are more sensitive to EPO than normal progenitors, and most homozygous progenitors are EPO independent. In this cohort of 306 JAK2V617F positive MPD patients, PV was associated with significantly lower platelet counts and higher hematocrit and granulocyte values than ET patient. The highest platelet count was associated with low JAK2V617F levels in PV, whereas high JAKV617F levels correlated with high hemoglobin and high granulocyte counts in ET [53]. Tefferi et al detected JAK2V617F in 75% of ET (n=60) and in 97% of PV patients (n=62), whereas allelic ratios exceeding 50% JAK2V617F indicating homozygosity were found in 70% of PV at diagnosis but never in ET [54]. Transition from heterozygosity to homozygosity for the JAK2V617F mutation represents a very important step in the progression from early to classic PV and subsequent post-PV and post- ET myelofibrosis (Table 5). JAK2V617F heterozygous and homozygous PV patients have increased haemoglobin, hematoctit and erythrocyte counts (> 6x1012/L) at time of diagnosis, experience increased incidence of pruritus, and a higher rate of fibrotic transformation as compared to ET. Sex appears to be a powerful genetic background modifier in JAK2V617F-positive MPDs as ET is more common in females and PV in males.
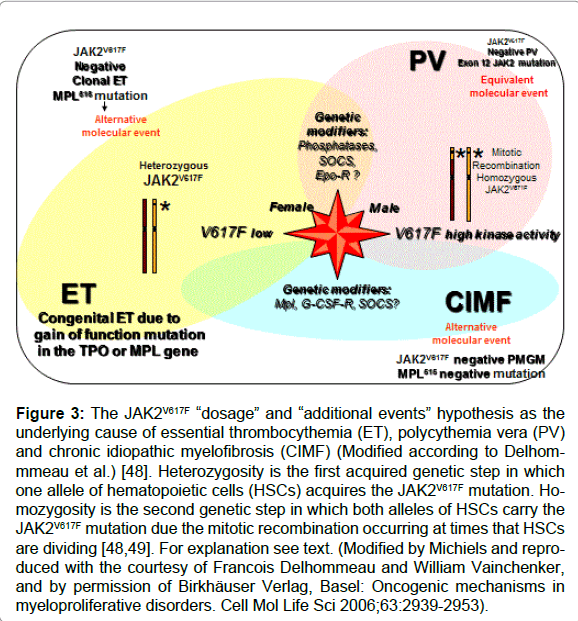
Figure 3: The JAK2V617F “dosage” and “additional events” hypothesis as the underlying cause of essential thrombocythemia (ET), polycythemia vera (PV) and chronic idiopathic myelofibrosis (CIMF) (Modified according to Delhommmeau et al.) [48]. Heterozygosity is the first acquired genetic step in which one allele of hematopoietic cells (HSCs) acquires the JAK2V617F mutation. Homozygosity is the second genetic step in which both alleles of HSCs carry the JAK2V617F mutation due the mitotic recombination occurring at times that HSCs are dividing [48,49]. For explanation see text. (Modified by Michiels and reproduced with the courtesy of Francois Delhommeau and William Vainchenker, and by permission of Birkhäuser Verlag, Basel: Oncogenic mechanisms in myeloproliferative disorders. Cell Mol Life Sci 2006;63:2939-2953).
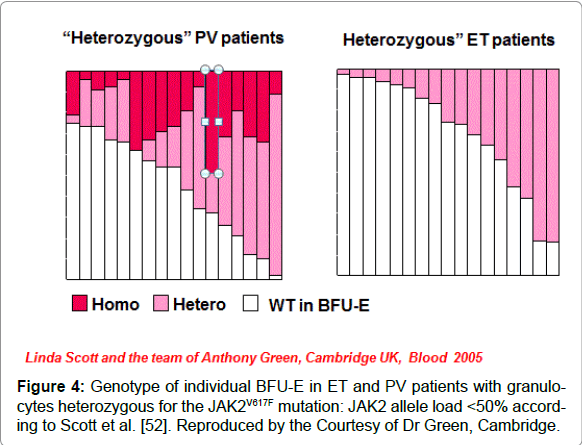
Figure 4: Genotype of individual BFU-E in ET and PV patients with granulocytes heterozygous for the JAK2V617F mutation: JAK2 allele load < 50% according to Scott et al. [52]. Reproduced by the Courtesy of Dr Green, Cambridge.
Mechanisms other than mitotic recombination such as duplication of the mutated allele is observed in a proportion of PV and MF patients displaying a gain of 9p, mostly due to trisomy 9 [55,56]. Campbell et al reported that the JAK2V617F mutation was associated with trisomy 9 with all 10 MPN patients investigated and was found in 28 of 29 MPN patients (PV, ET or MF) with a 20q deletion [57]. Scott et al identified JAK2 exon 12 mutations in 10 erythrocytosis patients with increased red cell mass but no JAK2V617F, of which according to PVSG criteria 6 could be diagnosed as PV and 4 as idiopathic erythrocytosis [58]. Pre-treatment bone marrow biopsies in 5 JAK2 exon 12 mutated PV patients showed a characteristic pattern of erythroid hyperplasia without morphological abnormalities of the megakaryocyte or granulocyte lineages [58]. Therefore, an overlap between “dosage” and “additional molecular events” hypotheses is very likely in patients with trilinear PV (Figure 3).
The JAK2 kinase activity in MPNs is dependent on the amount of heterozygous and homozygous JAK2V617F mutant protein, but also influenced by the various steps upstream or downstream the signalling pathways including MPL, JAK2, STAT-3 (Figure 2). This has been demonstrated in animal models overexpressing c-MPL [59]. MPL transgenic mice manifested with typical features of ET with a fourfold increase of platelet count, increased colony formation of megakaryocytes, and increase of clustered enlarged megakaryocytes in the bone marrow. The ET animals appeared healthy, had a very slight decrease of hematocrit (0.39 versus 0.42 in controls) despite an increase of bone marrow EEC, and survived normally with no evidence of myelofibrosis in the bone marrow [52]. The acquired MPLW515L and MPLW515K gain of function mutations have been discovered as the underlying etiology in ET patients (Figure 3) [60,61]. Screening of 1182 PVSG-defined MPD patients (318 ET, 242 PV, and 290 IMF) and 64 controls for MPL515 mutations resulted in the detection of MPL mutations either MPLW515L (n=17) or MPLW515K (n=5) in 20 MPN patients (MF in 4%, ET in 4=1%, and post-ET myelo fibrosis in 1), but not in the 242 PV patients and controls [60]. Six cases carried both MPLW515L and JAK2V617F alleles indicating that these alleles have functional complementation in MF (Figure 3). MPL515 mutated ET and MF represent a distinct entity of JAK2 wild type MPN without features of JAK2V617F mutated PV. These observations are in line with the “additional events” hypothesis, indicating that alternative or additional molecular abnormalities, like JAK2V617 mutation alone, combinations of JAKV617F and MPL515 mutations or other combinations of still unknown mutations contribute to distinct phenotypes of MPN.
Vannucchi et al studied 30 ET patients carrying the MPL515 mutation (18 MPLW515L and 12 MPLW515K), 9 males and 21 females, age 22-84 (mean 56) years [62]. The clinical presentation at diagnosis and follow-up was remarkable with a high incidence of major arterial event, 23%, venous thrombosis, 10%, microvessel disturbances, 60%, and major hemorrhage, 7%. The only abnormal laboratory finding was increased platelet counts, 956 + 331x109/L together with hemoglobin values in the lower range of normal (13.4 + 1.3 g/l), normal white blood cells (8.8 + 3.1x109/L), slight increase of LDH (459 + 182 U/L) and splenomegaly in only 5 (17%). Mutation allele burden was greater than 50% in half of MPLW515K patients compared to 17% of MPLW515L mutated ET patients. MPL515 and JAK2V617F mutations coexisted in 3 with MPLW515L and in 5 with MPLW515K allele mutations. As compared to the findings in ET with features of PV in blood (low serum EPO) and hypercellular bone marrow due to increased erythropoiesis (Figure 5) the general features of bone marrow reports cellularity in MPLW515L/K patients revealed significantly reduced erythropoiesis and decreased, which associated with increased number of clustered small and large to giant megakaryocytes and no significant increase in reticulin fibrosis in a normocellular bone marrow (Figure 6) without features of PV or PMGM [63-65]. In 2008 we discovered that JAK2/MPL wild type hypercellular ET associated with a PMGM bone marrow appeared to be the third distinct MPN entity (Figures 7 and 8).
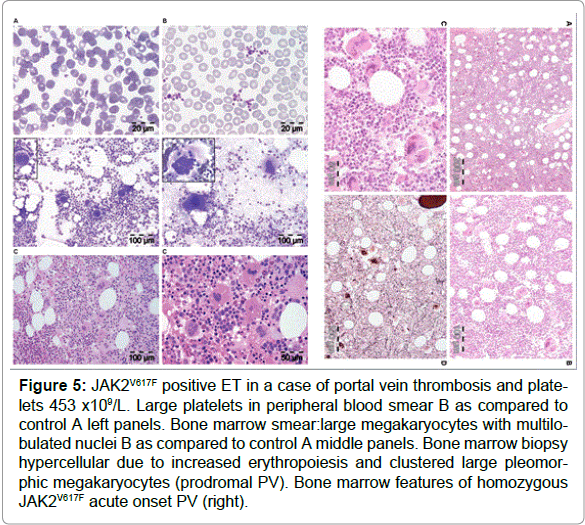
Figure 5: JAK2V617F positive ET in a case of portal vein thrombosis and platelets 453 x109/L. Large platelets in peripheral blood smear B as compared to control A left panels. Bone marrow smear:large megakaryocytes with multilobulated nuclei B as compared to control A middle panels. Bone marrow biopsy hypercellular due to increased erythropoiesis and clustered large pleomorphic megakaryocytes (prodromal PV). Bone marrow features of homozygous JAK2V617F acute onset PV (right).
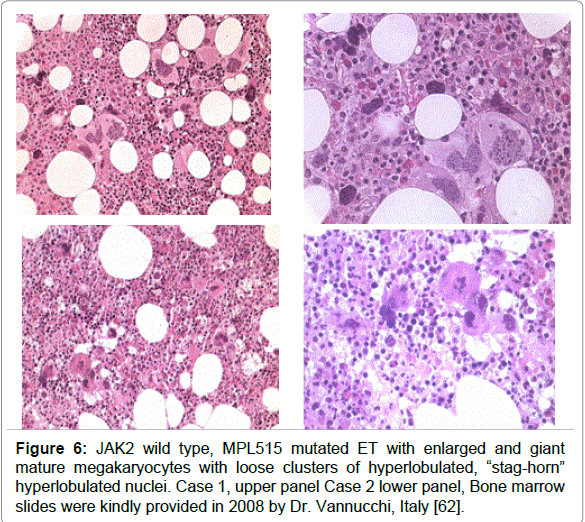
Figure 6: JAK2 wild type, MPL515 mutated ET with enlarged and giant mature megakaryocytes with loose clusters of hyperlobulated, “stag-horn” hyperlobulated nuclei. Case 1, upper panel Case 2 lower panel, Bone marrow slides were kindly provided in 2008 by Dr. Vannucchi, Italy [62].
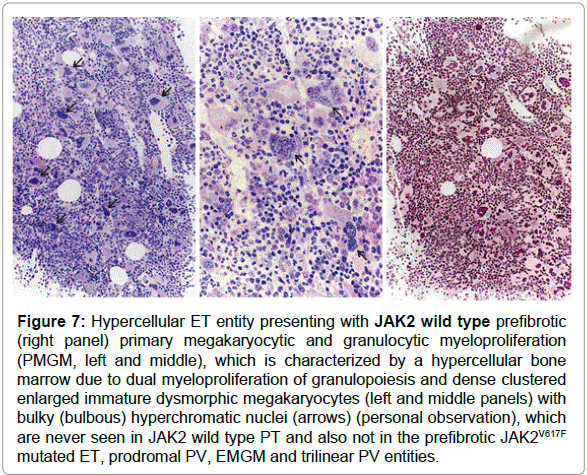
Figure 7: Hypercellular ET entity presenting with JAK2 wild type prefibrotic (right panel) primary megakaryocytic and granulocytic myeloproliferation (PMGM, left and middle), which is characterized by a hypercellular bone marrow due to dual myeloproliferation of granulopoiesis and dense clustered enlarged immature dysmorphic megakaryocytes (left and middle panels) with bulky (bulbous) hyperchromatic nuclei (arrows) (personal observation), which are never seen in JAK2 wild type PT and also not in the prefibrotic JAK2V617F mutated ET, prodromal PV, EMGM and trilinear PV entities.
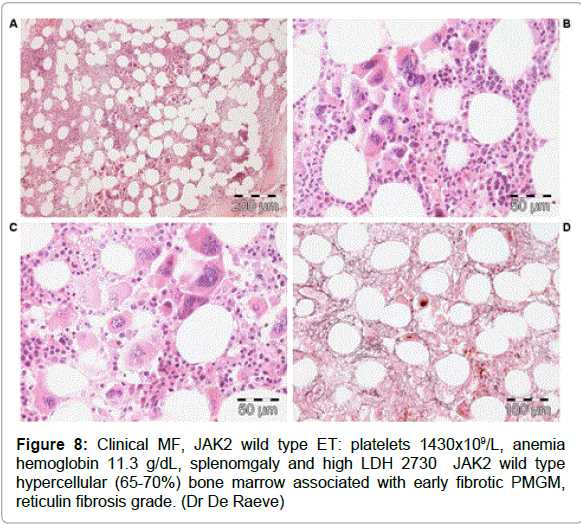
Figure 8: Clinical MF, JAK2 wild type ET: platelets 1430x109/L, anemia hemoglobin 11.3 g/dL, splenomgaly and high LDH 2730 JAK2 wild type hypercellular (65-70%) bone marrow associated with early fibrotic PMGM, reticulin fibrosis grade. (Dr De Raeve)
The 2008 ECMP criteria separate JAK2V617F mutated ET patients into three phenotypes of prefibotic MPNs at the bone marrow level: normocellular ET, early PV mimicking ET=prodromal PV (Figure 6) and ET with MGM (MF-0) bone marrow (EMGM) without features of leuko-erytroblastosis in the peripheral blood (Table 1). These three ECMP defined JAK2V617F mutated ET phenotypes do not differ significantly with regard to peripheral blood features, thrombocythemia related clinical presentation or laboratory findings and are to be treated equally based on clinical risk stratification for thrombotic and bleeding complications, irrespective of bone marrow features [51,63,64]. Pretreatment bone marrow biopsy will allow clinicians and pathologists to diagnose the early stages of thrombocythemia in various MPNs irrespective of JAK2V617F (Figure 5) or MPL515 mutation status (Figure 6). The 2008 ECMP criteria classify JAK2 mutated ET (Table 1) as: normocellular ET (Table 2); early PV mimicking ET (Table 1); prefibrotic ET associated with MGM (EMGM) with RF-0 or RF-1 and without features of leukoerythrocytosis and extramedullary hematopoiesis (Table 1); and post-ET MGM with MF-1, 2 and 3 and features of leucoerythroblastosis (Table 1). The 2008 ECMP criteria distinguish thrombocythemia in various MPNs from thrombocythemia associated with Ph1-chromosome and bcr/abl positive chronic myeloid leukemia (CML) [65] or Myelodysplastic Syndromes (MDS) including the so-called 5q-syndrome, which clearly differs from refractory anemia with ringed sideroblasts and significant thrombocytosis (RARS-T) [66- 69]. Among 9 RARS-T patients, 6 showed the presence of JAK2V617F mutation [68,69]. As compared to JAK2 wild type ET, JAK2V617F positive ET is characterized by higher values for hemoglobin, hematocrit, neutrophil counts, LAP score, by lower values for serum EPO levels, serum ferritin and MCV, and by increased cellularity of the bone marrow in biopsy material [65,66], indicating early thrombocythemic PV mimicking ET (“forme fruste” PV, stage 1 PV, Figure 5, Tables 2 and 4). JAK2 wild type ET patients represent a distinct category who had significantly higher platelet counts, normal serum EPO levels, a typical bone marrow picture of ET, no features of early PV, and are at lower risk for the development of thrombotic complications [70,71].
The detection of JAK2V617F in granulocytes with sensitive PCR techniques plays a key-role as a first intention diagnostic test for erythrocytosis, because it simplifies the diagnostic work-up of PV [72,73]. In the context of erythrocytosis (hematocrit >0.51 in males and >0.48 in females) the presence of the JAK2V617F mutation has a sensitivity of 95% and positive predictive value of 100% for the diagnosis of PV, and excludes congenital and secondary erythrocytosis. EEC and low serum EPO significantly contribute but are not sensitive enough to diagnose the broad spectrum of PV phenotypes [51]. Bone marrow histology assessment is a gold standard for the diagnosis of masked, overt and advanced JAK2V617F mutated and exon 12 mutated PV.
2007 and 2008 WHO defined Pimary Myelofibrosis (PMF) itself is not a disease because reticulin and collagen fibrosis are produced by polyclonal fibroblasts in response to cytokines released from the clonal granulocytic and megakaryocytic proliferative cells in both PV and MF (Table 3) [34,74]. Transformation to myelofibrosis is rare in ET and does occur in about one third of PV and in the majority of patients with ET associated with PMGM (MF-0) during long-term follow-up [22-24]. The grading of Reticulin Fibrosis (RF) was developed grading of myelofibrosis in bone marrow biopsies by pathologist (Table 3) [75]. A scoring system based on morphometric analysis (point intersection with an ocular grid) and quality of fibers (reticulin and collagen fibers) and the bone marrow fiber density (fine or course reticulin and some or course bundles of collagen) has been proposed by European consensus for grading of MF (Table 3) [34,75]. According to defined standardized semiquantitative grading of reticulin and collagen fibrosis in the bone marrow, MF can reliably be graded at the pathological bone marrow level as 0 in prefibrotic, as 1 in early fibrotic, as 2 in classical fibrotic and as 3 in classical sclerotic MF (Table 3) [34,75].
Spivak and his co-workers assessed the burden of JAK2V617F mutation and PVSG-defined MPD in 84 ET, 92 PV, and 19 fibrotic MF patients [76]. The JAK2V617F mutation was detected in 92% of PV, in 45% of ET, and in 42% of fibrotic MF patients. The median burden of JAK2V617F alleles was significantly lower ET (47%) than in PV (67%) in PV patients (p=<0.001) when stratified for disease duration, a JAK2V617F burden of 100% (homozygosity) was present in only 15% of PV less than three years from diagnosis compared to 40% of PV three to 10 years and 40% of PV between 10 and 26 years since diagnosis, but none in ET patients during very long-term followup (Figure 9) [76]. Passamonti et al studied the burden of JAK2V617F in WHO-defined MPD ET, PV, prefibrotic MF (p-MF), fibrotic MF (f-MF) and 16 post-PV myelofibrosis (Figure 10), [50]. The JAK2V617F mutation was detected in 92% of 25 PV, in 53% of 19 ET, in 58% of 12 MF-0 (ET associated with MGM) in 56% of 18 fibrotic MF, and in 100% of 16 post-PV myelofibrosis patients [50]. Interestingly, ET and p-MF patients had significantly lower percentage of mutated alleles than patients with PV (p=0.01), whereas patients with f-MF had much higher values than p-MF or ET (p=0.0008) (Figures 10). Circulating CD34 positive circulating cells were normal all patients with PV (N=25), ET (N=19) and p-CIMF (N=12) and 6 out of 18 f-MF patients had normal (<10x106/L) circulating CD34 cells. Conversely, 12 out of 18 f-MF and all post-PV MF [16] had increased CD34 circulating cells (Figure 10). These data indicate that ET and p-MF are not different at the molecular (JAK2V6217F) and biological (CD34 cells) level. Post-PV myelofibrosis had the highest percentages of mutant alleles approaching 100% homozygosity (Figure 8). PV and MF patients with a high mutation burden (granulocytes mutant alleles in excess of 50%) have leukocytosis, splenomegaly, increased LDH levels, increased circulating CD34-positive cells, a worse event free survival and a compromised overall survival as compared with those with lower mutation burden (granulocyte mutant alleles of 1-50%) mainly seen in ET and early stage PV [50].
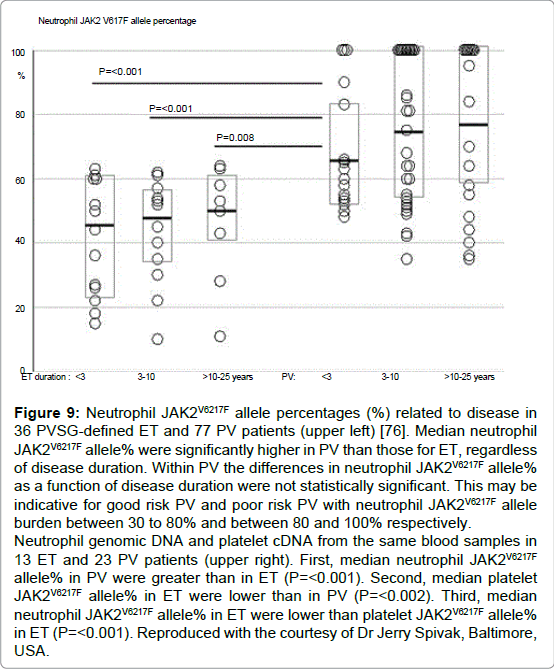
Figure 9: Neutrophil JAK2V6217F allele percentages (%) related to disease in 36 PVSG-defined ET and 77 PV patients (upper left) [76]. Median neutrophil JAK2V6217F allele% were significantly higher in PV than those for ET, regardless of disease duration. Within PV the differences in neutrophil JAK2V6217F allele% as a function of disease duration were not statistically significant. This may be indicative for good risk PV and poor risk PV with neutrophil JAK2V6217F allele burden between 30 to 80% and between 80 and 100% respectively. Neutrophil genomic DNA and platelet cDNA from the same blood samples in 13 ET and 23 PV patients (upper right). First, median neutrophil JAK2V6217F allele% in PV were greater than in ET (P=< 0.001). Second, median platelet JAK2V6217F allele% in ET were lower than in PV (P=< 0.002). Third, median neutrophil JAK2V6217F allele% in ET were lower than platelet JAK2V6217F allele% in ET (P=< 0.001). Reproduced with the courtesy of Dr Jerry Spivak, Baltimore, USA.
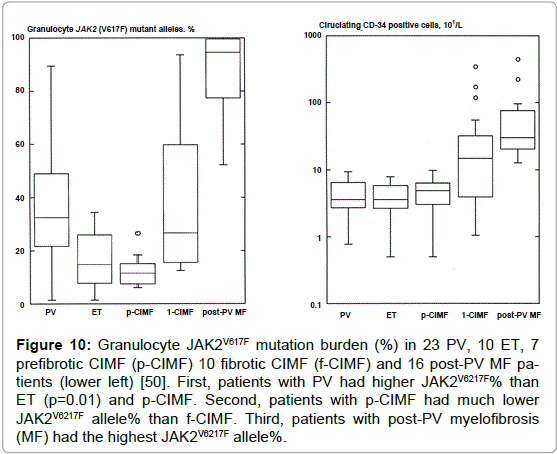
Figure 10: Granulocyte JAK2V617F mutation burden (%) in 23 PV, 10 ET, 7 prefibrotic CIMF (p-CIMF) 10 fibrotic CIMF (f-CIMF) and 16 post-PV MF patients (lower left) [50]. First, patients with PV had higher JAK2V6217F% than ET (p=0.01) and p-CIMF. Second, patients with p-CIMF had much lower JAK2V6217F allele% than f-CIMF. Third, patients with post-PV myelofibrosis (MF) had the highest JAK2V6217F allele%.
As compared to the 2007 → 2008 WHO classification [27,77], the use of the 2008 ECMP and the updated WHO-CMP [77-80] criteria clearly show that JAK2 wild type ET and MF lack specific PV laboratory and pathological features at diagnosis and during follow-up. This has been demonstrated for MPL515 mutated (ET/MF) (Figure 6) and for JAK2/MPL wild type hypercellular ET in PMGM (Figures 7 and 8). The flexible use of the 2008 ECMP → 2015 WHO-ECMP criteria should serve as pathognomonic diagnostic clues to each of the prefibrotic MPNs to distinguish early and overt PV MPN disease from primary or secondary erythrocytosis, and can be applied to document the natural history of myeloproliferative and fibrotic disease in JAK2V617F, MPL515 and JAK2 wild type ET and MF patients. The 2008 ECMP were the critical responses on the shortcomings of the 2007 WHO criteria and were conceptualised before the publication of the final 2008 WHO classification [36,37]. The 2008 ECMP and the updated 2014/2015 WHO-CMP detect and distinguish five distinct clonal MPNs caused by the JAK2V617F, exon 12 JAK2, MPL515 and CALR driver mutation leaving a small group of triple negative group of MPN [78-80].