Cell & Developmental Biology
Open Access
ISSN: 2168-9296
ISSN: 2168-9296
Review Article - (2013) Volume 2, Issue 1
Interstitial fibrosis is an end-stage pathophysiological feature of several clinically-important diseases involving the pulmonary, hepatorenal and cardiovascular systems and a common response to chronic injury, non-resolving inflammation or long-standing metabolic disease. Increased deposition of extracellular matrix (e.g., fibronectin, collagen), produced by persistently activated myofibroblasts, and attenuated matrix degradation results in disease progression. Disruption of normal tissue architecture eventually culminates in organ failure. Matrix turnover, in the context of both normal and abnormal wound repair, is regulated by several factors among which the most prominent ones are members of the transforming growth factor-β (TGF-β) superfamily. TGF-β1 stimulates fibronectin and collagen synthesis/accumulation at the wound site while impacting the extent and locale of plasmin/matrix metalloproteinaseinitiated stromal remodeling. This pericellular proteolytic cascade is highly-dependent on the generation of plasmin by urokinase (uPA) and tissue-type (tPA) plasminogen activators that, in turn, are subject to regulation by plasminogen activator inhibitor-1. In the normal response to injury, PAI-1 titrates the uPA/tPA/plasmin/MMP system to facilitate repair. Under pathologic conditions, excessive levels of PAI-1 at the site of injury suppress proteolytic activity promoting ECM accumulation and development of fibrosis. The continued clarification of mechanisms that control expression of disease-causative factors, such as PAI-1, will likely lead to the development of gene-specific therapies to inhibit the initiation and progression of the fibrotic process as well as to address the more difficult clinical challenge of reversal of established disease.
<The majority of fibrotic diseases are progressive, irreversible and eventually fatal; current treatments are limited and largely ineffective. Extent of interstitial involvement is a prognostic end-stage biomarker in chronic renal, hepatic and pulmonary disease particularly in the clinical setting of diabetes, hypertension, ischemia, obesity, metabolic syndrome, and acute injury. Regardless of etiology, elevated TGF-β1 levels (the predominant driver of the fibrotic response) and transcription of TGF-β1-responsive genes are closely linked to the activation of profibrotic signaling pathways. Among the most prominently involved genes, plasminogen activator inhibitor-1 (PAI- 1; SERPINE1(Serpin Peptidase Inhibitor, Clade E)), a member of the serine protease inhibitor family and the major negative regulator of the plasmin-based pericellular proteolytic cascade, is a causative factor in several clinically-significant fibrotic syndromes and healing anomalies [1-3]. Increased PAI-1 expression, due to increased TGF-β1 in the injured tissue, initiates and perpetuates the fibrotic response as part of an exuberant wound healing process. The translational potential in development of novel targeted approaches for the treatment of fibrosis highlights a need to further dissect the molecular mechanisms underlying TGF-β1-regulated PAI-1 gene control. Integration of basic molecular events underlying the etiology and progression of human fibrosis with in vivo interventional strategies and tissue repair outcomes assessments will provide the rationale for the clinical adaptation of focused therapies directed to the control of pathologically-relevant profibrotic genes.
PAI-1 overexpression is an important contributor to the pathophysiology of vascular and non-vascular fibrosis as well as a biomarker and a predictor of cardiovascular disease-associated mortality [4-7]. PAI-1 temporally/spatially regulates pericellular proteolysis and extracellular matrix accumulation impacting, thereby, stromal remodeling, inflammation, cell migration, proliferation and apoptosis. Most recently, a previously unknown cooperative network has emerged linking canonical (SMAD) and non-canonical (epidermal growth factor receptor (EGFR), RhoA GTPase) signaling [4] via TGF- β1-stimulated Src kinase activity [4,5]. The complexity of controls on the fibrotic process was highlighed by two major findings that defined a new paradigm of cross-talk between canonical and non-canonical TGF-β1-activated pathways required for expression of major fibrotic genes.
Cooperative canonical and non-canonical signaling initiated by TGF-β1 is required for PAI-1 transcription
This novel pathway of TGF-β1-induced PAI-1 expression requires pp60c-src-mediated EGFRY845 phosphorylation and MAP kinase activation that cooperate with TGF-βR-activated SMAD2/3 to transcribe the fibrotic PAI-1 and connective tissue growth factor (CTGF) genes [2,4,5,8,9]. TGF-β1-induced EGFR transactivation is significantly attenuated in Src-null cells [2,4,5] and PAI-1 induction is completely ablated in Src-deficient cells which have increased levels of the SMAD phosphatase PPM1A [5]. Transfection of a dominant-negative c-Src construct, furthermore, inhibited TGF-β1-induced PAI-1 expression while transfection of wild-type pp60c-src in Src-deficient cells “rescued” TGF-β1-mediated PAI-1 induction confirming participation of pp60csrc in PAI-1 gene control [2,4,5]. The EGFR-ERK and p38 pathways regulate phosphorylation and binding of upstream stimulatory factor (USF2), a basic helix-loop-helix-leucine zipper (bHLH-LZ) MYC-like protein, to critical E box motifs (CACGTG) in the PE1 and PE2 sites in the PAI-1 promoter while RhoA/p160 ROCK control PPM1A levels and the duration of SMAD2/3 phosphorylation [5]. Replacement of repressive USF1 homodimers with stimulatory USF1/2 heterodimers or USF2 homodimers was required for transcriptional activation of the PAI-1 gene [10]. TGF-β1-induced PAI-1 expression, therefore, involves SMAD2/3 activation and a repressor-to-activator subtype switch (USF1→USF2) at the PE2 E box [8,9]. Consistent with this model, USF2, but not USF1, levels are increased in the fibrotic kidney [2]. SMAD2/3 and USF2 occupy closely juxtaposed binding motifs at the TGF-β1-responsive PE2 region E box in the human PAI-1 promoter [8,10,11]. Both elements are essential for PAI-1 induction. SMAD3 knockdown or use of SIS3, a specific inhibitor of SMAD3 phosphorylation, as well as dominant-negative interference with USF DNA-binding or introduction of PE2 region deoxyoligonucleotide decoys inhibit TGF-β1-mediated PAI-1 transcription [4,10,11]. Interference with EGFR/pp60c-src/MAP kinase/p160ROCK activation or USF function, moreover, attenuate PAI-1 synthesis. These data underscore the importance of USF and cooperative TGF-β1/EGFRRho/ ROCK pathways (Figure 1) in control of genes that regulate the tissue response to injury and subsequent fibrosis.
The pp60c-src kinase targets focal adhesion kinase (FAK) and caveolin-1: a pathway to control SMAD2/3 phosphorylation
TGF-β1 stimulates FAKY397, Y577, Y861 and caveolin-1Y14 phosphorylation and both FAK and caveolin-1 are required for TGF- β1-induced PAI-1 expression [5]. While FAK autophosphorylation at Y397 is Src-independent [12], TGF-β1 failed to phosphorylate FAK Y577, Y86 in Src-/- fibroblasts confirming a role for Src kinases in FAK activation in response to TGF-β1. FAK is critical, moreover, for both TGF-β1-induced PAI-1 and CTGF expression as neither are detectable in FAK-null cells [5]. FAK deficiency also affected other TGF-β1 signal intermediates. SMAD3 activation in TGF-β1-treated FAK-/- cells is substantially reduced compared to wild-type controls impacting the control of pSMADs involved in PAI-1 expression [4,5]. Indeed, the TGF-β1-dependent increase in caveolin-1Y14 phosphorylation (a Src kinase site) was similarly attenuated by FAK deficiency while caveolin- 1Y14 phosphorylation was undetectable in Src-/- cells [5]. This has consequences on TGF-β1 signaling since caveolin-1-/- cells are deficient in PAI-1 induction compared to caveolin-1+/+ cells. TGF-β1-stimulated caveolin-1Y14 phosphorylation, c-SrcY416 activation and subsequent PAI-1 expression were, in fact, completely eliminated by the Src kinase inhibitor SU6656 [4,5]. The level and time course of TGF-β1- stimulated SMAD2/3 activation were both found to be decreased in caveolin-1-/- as well as Src-/- or FAK-/- cells [5]. Stable re-introduction of a wild-type caveolin-1 construct in caveolin-1-/- cells rescued PAI-1 expression while siRNA knockdown of caveolin-1 effectively suppressed TGF-β1-stimulated PAI-1 induction confirming a role of caveolin-1 in TGF-β1 signaling. The importance of this pathway in PAI-1 gene control is underscored by findings that TGF-β1-initiated RhoA activation is significantly reduced in caveolin-1-deficient cells and an intact Rho pathway is a critical negative regulator of PPM1A, a SMAD2/3 phosphatase [5]. SMAD2/3 activation and subsequent PAI- 1 induction by TGF-β1 is, in fact, suppressed by genetic ablation of caveolin-1. These data collectively implicate caveolin-1 as an upstream regulator of RhoA-ROCK-SMAD2/3 signaling leading to control of PPM1A activity impacting downstream PAI-1 expression.
From molecular mechanisms to clinical utility
TGF-β1 drives formation of p53-SMAD2/3 complexes while RhoA/ROCK activation represses the SMAD phosphatase PPM1A maintaining the phospho-SMAD2/3 dependency of PAI-1 transcription. The EGFR is a direct upstream regulator of MEK/ ERK1/2 while Rho/ROCK modulates both the duration of SMAD2/3 phosphorylation (via PTEN/PPM1A) and nuclear accumulation. E-box motifs (CACGTG) in the PE1/PE2 promoter regions of the human PAI-1 gene are platforms for a MAP kinase-directed USF subtype switch (USF-1→USF-2) in response to growth factor addition suggesting that the EGFR→MEK/ERK axis impacts PAI-1 expression, at least partly, through USF-dependent transcriptional controls (Figure 1). The continued clarification of mechanisms that control expression of disease-causative factors (e.g., PAI-1) will likely lead to the development of gene-specific therapies to inhibit the initiation and progression of the fibrotic process as well as to address the more difficult clinical challenge of reversal of established disease. The identification of fundamental controls on PAI-1 transcription may lead to new targeted, perhaps combinatorial, therapies and clinically-relevant options for the treatment of fibrotic disorders in which PAI-1 dysregulation is a major underlying pathogenic feature. Pharmacologic agents that disrupt Src and MAP kinase signaling or p53 pathways are already in clinical trials and small molecule PAI-1 functional inhibitors are in the pre-clinical test stage [7]. Optimization of such approaches may well lead to the eventual realization of the practical utility of transcription-focused drugs.
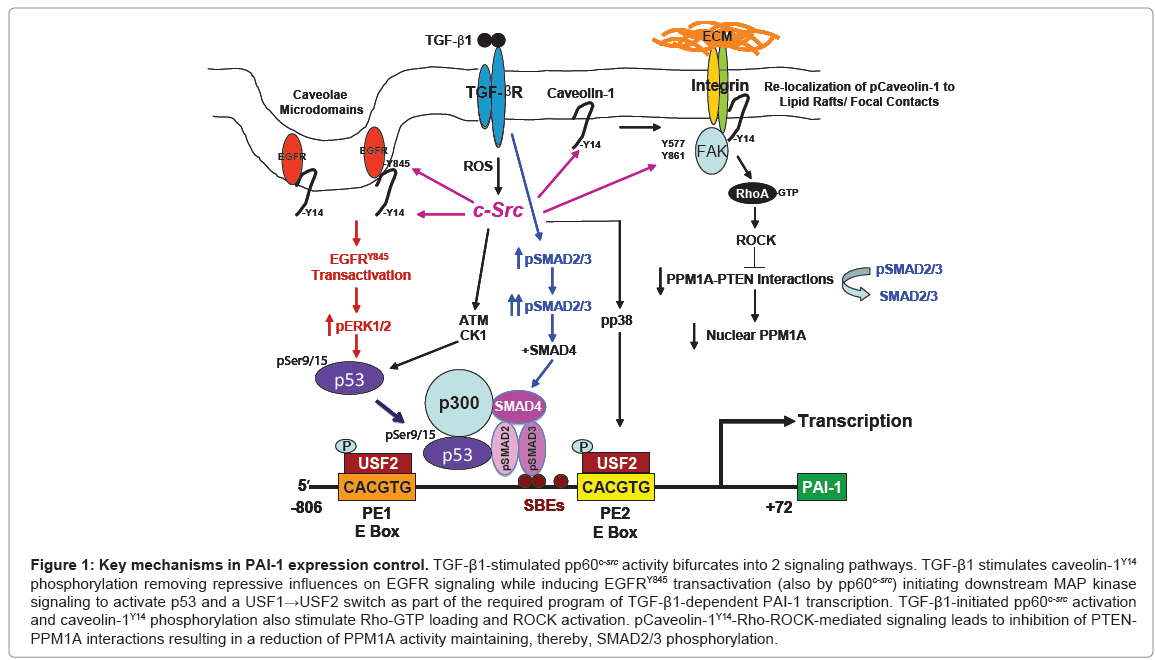
Figure 1: Key mechanisms in PAI-1 expression control. TGF-β1-stimulated pp60c-src activity bifurcates into 2 signaling pathways. TGF-β1 stimulates caveolin-1Y14 phosphorylation removing repressive influences on EGFR signaling while inducing EGFRY845 transactivation (also by pp60c-src) initiating downstream MAP kinase signaling to activate p53 and a USF1→USF2 switch as part of the required program of TGF-β1-dependent PAI-1 transcription. TGF-β1-initiated pp60c-src activation and caveolin-1Y14 phosphorylation also stimulate Rho-GTP loading and ROCK activation. pCaveolin-1Y14-Rho-ROCK-mediated signaling leads to inhibition of PTENPPM1A interactions resulting in a reduction of PPM1A activity maintaining, thereby, SMAD2/3 phosphorylation.
We would like to acknowledge NIH grant GM057242 and the Community Foundation of Sarasota County, Inc. from the Charlotte P. Graver Fund.