Journal of Clinical Toxicology
Open Access
ISSN: 2161-0495
ISSN: 2161-0495
Research Article - (2022)Volume 12, Issue 2
There studies centering the potential usage of benzensulfonamide derivatives as anticancer agents are still limited. So, in this study, a series of new sulfonamide drugs were synthesized by the reaction of aldehydes thiosemi carbazones derivatives with benzene sulphonyl chloride to form benzylidene-N-(phenylsulfonyl) hydrazine-1-carbothioamide derivatives. Spectral techniques like Fourier Transformer Infrared Analysis (FTIR), Proton Nuclear Magnetic Resonance (1HNMR) and Mass spectroscopy were used to characterize the newly synthesized compounds. The two derivatives, 2-benzylidene-N-(phenylsulfonyl) hydrazine-1-carbothioamide (4a) and 2-(4-chloro benzylidene)-N-(phenylsulfonyl) hydrazine-1-carbothioamide (4d) exhibited the most potent anticancer effects against MCF-7 breast carcinoma cell lines. Meanwhile, these two derivatives showed the lowest antioxidant activities. To study the study of anti-breast cancer activity of the newly synthesized compounds, molecular docking study were used to analyze the binding energy for the non-bonding interactions between the ligand (studied compounds) and receptor (4PYP (pdb code: 4FA2) against human breast cancer (MCF-7) cells. The bioavailability of all studied compounds was confirmed by pharmacological investigations using molinspiration and ADMET online servers.
Benzenesulfonamids; Anticancer activity; Antioxidant activity; Molecular docking; ADMET
Human cancer is a serious threat to the world public health as it is the second leading cause of death in humans after cardiovascular diseases[1]. Although there are several human cancer treatment strategies, such as radiation therapy and chemotherapy, in most cases, drug resistance and high systemic toxicity limit the effectiveness of successful treatment.
In women worldwide, Breast cancer is second most common cause of death and one of the oldest known forms of human cancer [2]. Cyclophosphamide plus doxorubicin cause several abnormal side effects in normal cells although they are one of the most common drugs that treat breast cancer [3]. Due to the multidrug resistance and unfavorable side effects of current cancer chemotherapeutic, a realistic need for development of new anti-cancer drugs has been arisen. These drugs including synthetic compounds, with less multidrug resistance of tumor cells and limited toxicity to normal tissue [4].
Sulfonamides are organic compounds with functional group of sulfamoyl SO2-NH moiety that is the basis of several groups of sulfa drugs [5]. Also, in synthetic chemistry, both benzenesulfonamides and p-toluenesulfonamide have been widely explored; However, limited work has been performed on the α-tolylsulfonamide [5]. In recent years, sulfonamides have been found to be associated with antibacterial, anticancer, antiviral, anti-inflammatory, carbonic anhydrase inhibitory [6-10], antihypertensive, antimalarial, antitumor, and antiprotozoal activities [11-14]. Moreover, some benzenesulfonamide derivatives also showed broad spectrum of biological activities such as carbonic anhydrase inhibitors [15], herbicides and plant growth regulators, elastase inhibitors and Clostridium histolyticum collogenase inhibitors [16-18]. Further, benzenesulfonamide derivatives are currently used as anticancer agents against different breast cancer cell lines such as MCF-7, lung cancer cell (A549), prostate cancer cell (Du-145) and cervical cells (HeLa) [19].
ROS or Reactive Oxygen Species like hydroxyl radical, superoxide anion, and hydrogen peroxide attack several bioactive molecules such as DNA, enzymes, proteins and other biomolecules. Metabolic disorders, inflammatory, cancer and cellular aging are caused by Oxidative stress conditions. ROS and free radicals induce oxidative damage that could be inhibited by Antioxidants. So, antioxidant treatment therapy has also acquired enormous significance in the healing and treatment of cancer. Such elucidation needs to be convinced and elucidated.
Molecular docking turns out to be one of the most important tools to figure the binding affinity and how the ligands (the new synthesized molecules) impede the target, which is helpful in the optimization of the most promising compounds that would have the highest biological activity. The estimation of Absorption, Distribution, Metabolism, Excretion and Toxicity properties (ADMET properties) of the tested compounds is a crucial test in the field of drug development. In silico drug design methods, toxicity potential and pharmacokinetic parameters are commanding tools for the design and the study of the toxic and physicochemical properties of the novel synthesized compounds as well as discovering their interaction with target receptors. To rapid authentication of promising compounds for preclinical studies and clinical trials [20].
In the light of the wide range of medical applications of sulfonamide derivatives as a new anticancer molecule, researchers were encouraged to synthesize and design of innovative drugs containing sulfonamide compounds. Thus, we hereby report the preparation, characterization, antioxidant and anticancer activities of some novel benzenesulfonamide derivatives. Also, we calculated the binding energy for the non-bonding interactions between the ligand (studied compounds) and receptor (4PYP (pdb code: 4FA2) using molecular docking to study anti-breast cancer activity of the newly synthesized compounds against human breast cancer (MCF- 7) cells. ADMET studies were performed to compute the toxicity risks and drug-relevant properties of the studied compounds.
MCF7 as Breast carcinoma cell line were obtained from ATCC (the American type culture collection), Minisota, and USA have been used in this study. RPMI-1640 medium, Dimethylsulphoxide (DMSO), Fetal Bovine Serum, trypan blue, Trypsin-EDTA and Penicillin/Streptomycin antibiotic were purchased from Sigma Aldrich Chemical Co. Tris buffer purchased from Applichem, Germany. Reagents and starting materials were used as purchased from commercial suppliers without further purification.
General procedure for synthesis of 2-(4-substitutedbenzylidene)- N-(phenylsulfonyl)hydrazine-1- carbothioamide (4a-4e)
50 mL of 10 mmol solution of 2-(4-substitutedbenzylidene) hydrazine-1-carbothioamide (2a-2e) in dichloromethane was added drop wise to 20 mL of a stirred mixture of 10 mmol benzene sulfonyl chloride and 14 mmol sodium carbonate in dichloromethane. After stirring the resultant reaction mixture overnight at room temperature, 20 mL distilled water was added to it. The yielded solids were filtered and washed several times with distilled water and crystallized to gives compounds (4a-4e). The washed organic solid product was dried over anhydrous sodium sulfate under reduced pressure. To ensure the high purity of all the prepared compounds, recrystallization was done for further purification.
Characterization of the newly synthesized samples
The spectral data together with the physical constants of all of the previously prepared compounds were identified as follows. IR spectra were recorded using a Fourier Transform Infrared (FTIR) spectrometer (VERTEX 70 FT-IR). On a Varian 400 MHz spectrometer, 1H-NMR (Proton Nuclear Magnetic Resonance) spectra were determined for solutions in (CD3)2SO. TMS was used as an internal standard. Varian MAT 112 spectrometer was used to record the mass spectra of the prepared compounds. All melting points are uncorrected and elucidated using Stuart Scientific Melting Point (SMP) apparatus (USA).
Anti-cancer activity
Cultured MCF-7 human breast cancer cell was maintained in RPMI- 1640 medium supplemented with 10% Fetal Bovine Serum (FBS), 100 units/ml Penicillin 2 mg/ml Streptomycin at a temperature of 37°C. Cells were cultured in T25 nunclon sterile tissue culture flasks, and sub-fused cells were separated in calcium-free phosphate buffer with a 0.25% trypsin-EDTA solution containing 2.5 g of porcine trypsin and counted in a hemocytometer.
To calculate the cell count/ml of cell suspension, 50 μl of 0.05 % trypan blue solution was added to 50 μl of the single cell suspension and the non-stained (viable) cells were counted and we calculate the cell count/ml using the following equation.
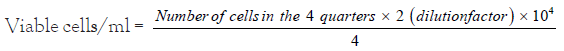
The cells were then diluted to give the required cell number for each experiment. The potential cytotoxicity of drug on human cancer cell line was determined using Vichai method. Where the cells were plated at 5 × 103 cells per well of 96-well plates in in a 150 µl of growth medium (RPMI-1640) and left for 24 hours to attach to the plates then the cells cultured for 24 h with various concentrations (0, 5, 12.5, 25, 50 µg/ml ) of studied compounds (Derivatives were prepared by dissolving 1: 1 Stock solution and stored at -20°C in Dimethylsulfoxide (DMSO) at 100 mM). The cells were fixed with 50 μl 10% final concentration of cold trichloroacetic acid for 1 hour at 4°C. Then washed with distilled water and stained with 50 μl 0.4% Sulphorhodamine-B (SRB) dissolved in 1% acetic acid for 30 minutes at room temperature. The plates were washed with 1% acetic acid and air-dried. The dye was solubilized with 100 μl/well of 10 M tris base (pH 10.5) and Optical Density (OD) of each well was measured spectrophotometrically at 570 nm. The experiment was repeated 3 times.
The survival percentage was calculated as follows:
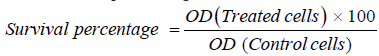
The IC50 values (the concentrations of drug required to produce 50% inhibition of cell growth) were also calculated.
DPPH radical scavenging activity
After minor modifications, Liyana-Pathiranan, et al. [20], was used to estimate the newly synthesized organic compounds effect of the on DPPH radical where 15 µl of varying concentrations of the newly synthesized organic compounds (0.25, 0.5, 1, 2 mg/ml) was added to 185 µl 0.135 mM DPPH in methanol, in a 96-well plate. The reaction mixture was then vortexed and left in the dark at room temperature for 30 minutes. By using a microplate reader, the absorbance of the mixture was measured at 570 nm. Trolox is used as an antioxidant reference compound. The DPPH free radical scavenging ability was calculated using the following formula:
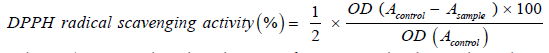
Where Acontrol is the absorbance of DPPH radical in ethanol and, Asample is the absorbance of sample of fabricated organic derivative + DPPH radical dissolved in ethanol.
Molecular docking
The studied receptor structure 4PYP (pdb code: 4FA2) was downloaded from the pdb (protein data bank) (http://www.rcsb.org/) and the energy was minimized before docking. Then we calculated the binding energy for the non-bonding interactions between the ligand (studied compounds (4a-4e)) and receptor (PD: 4FA2), in addition to that we also reported important residues that stabilize the ligand (the synthesized compounds) at the active site of the receptor. In the current study, molecular docking was proposed to infer the required receptor-ligand binding site and its binding affinity. Initially, discovery studio was used to remove eutectic ligand cofactors and water from proteins. Auto Dock Tools (ADT) graphical user interface for performing docking between ligands and receptors [21]. Subsequently, by the Kollman and Gasteiger method, the atomic charge was calculated after polar hydrogen attachment. The active site of the protein is defined by the grid size of 60Å × 60Å × 60Å, and the process is performed using Lamarckian Genetic Algorithm (LGA). Discovery Studio Accelrys software (DS) was used to evaluate the ADMET properties of the studied compounds. DS provides a method for assessing the disposal and potential toxicity of ligands in organisms. ADMET properties were analyzed and calculated using the ADMET protocol in DS that contains published models that could be used to calculate and analyze ADMET characteristics. In addition, ligands that are unlikely to be drugs, like unsuitable lead for example, were removed by DS that apply restricted rules to remove such ligands. These rules based on the presence or absence and frequency of certain chemical groups. Six mathematical models are used by this module to quantitatively predict ADMET properties through a conventional set of rules that identify the threshold ADMET features of the molecular chemical structure based on the availability of drug information.
The spectral data and physical constants of the newly synthesized compounds. 2-benzylidene-N-(phenylsulfonyl) hydrazine-1-carbothioamide (4a)
Yield: 78%; melting point 133°C-138°C, IR representative peaks appear at (umax/cm-1 (KBr)): 3274, 3097, 1644, 1328, 1267 and 1149. 1H-NMR (CDCl3) representative peaks appear at: δ=10.9 (br, s, 1H, CS-NH-SO2), 9.19 (s, 1H, N-NH-CS), 8.15 (s, 1H, CH=N), 7.21-7.77 (m, 10H, CH-aromatic), m/z 319 for C15H15N3O2S2. From the elemental analysis data, analysis calculated for C15H15N3O2S2 were C, 52.65; H, 4.10; N, 13.16; S, 20.08; O, 10.02 and the analysis found for the compound are: C, 51.95; H, 4.02; N, 13.09; S, 19.77; O, 9.86.
2-(4-methyl benzylidene)-N-(phenylsulfonyl)hydrazine-1- carbothioamide (4b)
Pale yellow, Yield: 69%; mp 140°C-142°C, IR representative peaks appear at (umax/cm-1 (KBr)) 3298, 3089, 1653, 1335, 1250, 1135. 1H-NMR (CDcl3) representative peaks appear at: δ=11.1 (br, s, 1H, CS-NH-SO2), 9.43 (s, 1H, N-NH-CS), 8.23 (s, 1H, CH=N), 7.34- 7.80 (m, 9H, aromatic protons), 2.56 (s, 3H, CH3), m/z 333 for C14H13N3O2S2. From the elemental analysis data, analysis calculated for C14H15N3O2S2 were C, 54.03; H, 4.53; N, 12.60; S, 19.23; O, 9.60 and the analysis found for the compound are: C, 53.75; H, 4.32; N, 12.45; S, 19.09; O, 9.55.
2-(4-methoxy benzylidene)-N-(phenylsulfonyl)hydrazine-1- carbothioamide (4c)
Yellow, Yield: 65%; mp 153°C–154°C, IR representative peaks appear at (umax/cm-1 (KBr)) 3289, 3083, 1643, 1324, 1264, 1160, 1130. 1H NMR (CDcl3) representative peaks appear at: δ=11.3 (br, s, 1H, CS-NH-SO2), 9.56 (s, 1H, N-NH-CS), 8.29 (s, 1H, CH=N), 7.01-7.99 (m, 9H, aromatic protons), 3.99 (s, 3H, OCH3), m/ z=349 for C15H15N3O3S2. From the elemental analysis data, analysis calculated for C15H15N3O3S2 were C, 51.56; H, 4.33; N, 12.03; S, 18.35; O, 13.74 and the analysis found for the compound are: C, 51.39; H, 4.11; N, 11.95; S, 18.30; O, 13.59.
2-(4-chloro benzylidene)-N-(phenylsulfonyl)hydrazine-1- carbothioamide (4d)
Light brown, Yield: 67%; mp 165°C-167°C, IR representative peaks appear at (umax/cm-1 (KBr)) 3296, 3090, 1647, 1333, 1257, 1153, 811. 1H NMR (CDcl3) representative peaks appear at: δ=11.25 (br, s, 1H, CS-NH-SO2), 9.38 (s, 1H, N-NH-CS), 8.44 (s, 1H, CH=N), 7.39-7.87 (m, 9H, aromatic protons), m/z=353 for C14H12N3O2S2Cl. From the elemental analysis data, analysis calculated for C14H12N3O2S2Cl were C, 47.52; H, 3.42; N, 11.88; S, 18.12; Cl, 10.02; O, 9.04 and the analysis found for the compound are: C, 47.32; H, 3.35; N, 11.77; S, 18.01; Cl, 9.89; O, 8.85.
2-(4-dimethylamino)benzylidene)-N-(phenylsulfonyl) hydrazine-1-carbothioamide (4e)
Yellow, Yield: 62%; mp 160°C-162°C, IR representative peaks appear at (umax/cm-1 (KBr)) 3294, 3095, 1652, 1329, 1244, 1150. 1H NMR (CDCl3) representative peaks appear at: δ=10.89 (br, s, 1H, CS-NH-SO2), 9.50 (s, 1H, N-NH-CS), 8.65 (s, 1H, CH=N), 7.26-7.97 (m, 9H, aromatic protons), 2.95 (s, 6H, CH3), m/z=353 for C16H18N4O2S2. From the elemental analysis data, analysis calculated for C16H18N4O2S2 were C, 53.02; H, 5.01; N, 15.46; S, 17.69; O, 8.83 and the analysis found for the compound are: C, 53.17; H, 5.09; N, 15.39; S, 17.61; O, 8.90.
Samples characterization
From the above mentioned spectral data and physical constants of the newly synthesized compounds we concluded that: Formation of aldehydes thiosemicarbazide is the key reaction in our plan to produce new 2-(4-substituted-benzylidene)-N-(phenylsulfonyl) hydrazine-1-carbothioamide (4a-4e) as sulphanamide drugs by the reaction of 2-(4-substituted-benzylidene) hydrazine-1- carbothioamide (2a-2e) with benzene sulphonyl chloride (3) in dichloromethane in presence of Sodium carbonate. The sulfnoamide derivatives were readily accomplished in good yield, 78%, 69%, 65%, 67%, and 70% for derivatives, respectively. All derivatives were confirmed and established by elemental analysis and shown characteristic peaks for sulfonamide drugs in FT-IR umax/ cm-1 (KBr) 3274, 3298,3289,3296,3277 (NH (4a-4e) respectively), 3097, 3089, 3083,3090, 3081 (C-H SP24 respectively), 1644, 1653, 1643, 1647, 1649 (C=C (4a-4e) respectively) and 1247,1250, 1264, 1257, 1255 (C=S (4a-4e) respectively), also characteristic peaks in 1H-NMR (CDcl3): δ=10.9 (br, s, 1H, CS-NH-SO2), 9.19 (s, 1H, N-NH-CS), 8.15 (s, 1H, CH=N), 7.21-7.77 (m, 10H, aromatic protons), for compound 4a, δ=11.1 (br, s, 1H, CS-NH-SO2), 9.43 (s, 1H, N-NH-CS), 8.23 (s, 1H, CH=N), 7.34-7.80 (m, 9H, aromatic protons), 2.56 (s, 3H, CH3) for compound 4b, δ = 11.3 (br, s, 1H, CS-NH-SO2), 9.56 (s, 1H, N-NH-CS), 8.29 (s, 1H, CH=N), 7.01- 7.99 (m, 9H, aromatic protons), 3.99 (s, 3H, OCH3) for compound 4c, δ=11.25 (br, s, 1H, CS-NH-SO2), 9.38 (s, 1H, N-NH-CS), 8.44 (s, 1H, CH=N), 7.39-7.87 (m, 9H, aromatic protons) for compound 4d and δ=10.89 (br, s, 1H, CS-NH-SO2), 9.50 (s, 1H, N-NH-CS), 8.65 (s, 1H, CH=N), 7.26-7.97 (m, 9H, aromatic protons), 2.95 (s, 6H, CH3) for compound 4e.
Anti-cancer activity
The compound 4e showed the most potent anticancer activities against MCF-7 breast carcinoma cell lines since it has the lowest IC50 of 28.2 µg/ml. In spite of its higher potent effect, its anticancer effects are still lower than doxorubicin (standard anticancer drug) (IC50: 12.80 µg/ml). The four other derivatives 4a-4d exhibited less anticancer activities and have IC50 of 38.1, 38, 37.8 and 41 µg/ml respectively as shown in Table 1 and Figures 1 and 2.
| Derivatives | IC50 (µg/ml) |
|---|---|
| Doxorubicin (standard anticancer drug) | 12.8 |
| (4a) | 38.1 |
| (4b) | 38 |
| (4c) | 37.8 |
| (4d) | 41 |
| (4e) | 28.2 |
Table 1: In vitro anticancer activity of all synthesized compounds on MCF-7 cell line.

Figure 1: Synthesis of new 2-(4-substituted-benzylidene)-N- (phenylsulfonyl)hydrazine-1-carbothioamide (4a-4e).

Figure 2: The survival percent and IC50 of the newly synthesized compounds against MCF-7 cell line.
Antioxidant activity
The DPPH radical scavenging activity percent of sulfonamide derivatives, in comparison with that of the reference antioxidant gallic acid, is represented in Table 2. All tested derivative exhibited antioxidant activity. The derivative 2-(4-methoxy benzylidene)- N-(phenylsulfonyl)hydrazine-1-carbothioamide (4c) showed the highest antioxidant activity at concentration of 0.25, 0.5 and 1 mg/ml while the derivative 2-benzylidene-N-(phenylsulfonyl) hydrazine-1-carbothioamide (4a) has the less potent effect. Derivative 2-(4-methoxy benzylidene)-N-(phenylsulfonyl)hydrazine- 1-carbothioamide (4c) is more effective in scavenging free radicals than the reference antioxidant, gallic acid at 0.5 mg/ml while it has comparable effect to the reference antioxidant at 0.25 and 1 mg/ml. The two derivatives 2-benzylidene-N-(phenylsulfonyl) hydrazine-1-carbothioamide (4a) and 2-(4-chloro benzylidene)-N- (phenylsulfonyl)hydrazine-1-carbothioamide (4d) showed the lowest antioxidant activities. Derivative 2-(4-(dimethylamino)benzylidene)- N-(phenylsulfonyl)hydrazine-1-carbothioamide (4e) has similar activity to the reference antioxidant at such concentration 0.25 mg/ml.
| Derivatives | 5 mg/ml | mg/ml | g/ml |
|---|---|---|---|
| Gallic acid (standard antioxidant) | 3.327 ± 1.753 g | 9.218 ± 0.283 f | 3.012 ± 1.133 e |
| (4a) | 8.289 ± 0.283 r | 8.254 ± 0.055 p | 9.524 ± 0.340 m |
| (4b) | 0.802 ± 0.227 o | 3.318 ± 0.622 l | 8.380 ± 0.168 j |
| (4c) | 2.536 ± 0.196 g | 5.447 ± 0.168 cd | 4.144 ± 0.113 de |
| (4d) | 8.539 ± 5.436 p | 9.161 ± 3.227 op | 0.713 ± 0.058 m |
| (4e) | 1.630 ± 0.171 g | 8.630 ± 0.284 h | 2.989 ± 0.168 g |
Note: The data are represented as mean ± SE of three replicates for each concentrations. The means, which have different superscript letters, are significantly different at p<0.05.
Table 2: In vitro antioxidant activity of all synthesized compounds.
Molecular docking
Based on the reported results on the biological activity of p38a MAP kinase in fighting breast cancer and the activity of different functionalized chromone derivatives as p38a inhibitors, all of our newly synthesized compounds were fabricated to introduce new p38a MAP kinase inhibitors for human breast cancer MCF-7 cells [22]. The ligand-receptor intermolecular interactions between the studied compounds are also investigated. The docking simulation process for each synthesized compound was done and best conformation is chosen to be the compound with a larger negative binding energy value. The 3D and 2D structures of the ligandreceptor structures of all studied compounds are (4a-4e). The estimated binding energies, inhibition constant, intermolecular energies and reference RMSD produced from docking for all studied compounds are displayed in Table 3. All studied compounds formed stable complexes with receptor with high binding energy. Our study revealed that among the novel synthesized compounds, compound 4e exhibited the best docking energy (the highest binding energy) with binding affinity of -10.48 kcal/mol followed by compound 4c (-10.26 kcal/mol) (Table 3). This is in a good agreement with the data obtained from the calculated IC50 values (Table 1) which shows that compound 4e has the highest activity (the lowest IC50 value=28.2 µg/ml) followed with compound 4c (IC50 value=37.8 µg/ml). Therefore, the compounds studied, especially compounds 4e and 4c, can be used as potential compounds against (MCF-7 cells) human breast cancer.
| Compounds | Binding energy (k cal/mol) | Estimated inhibition constant (µm) | Intermolecular energy (k cal/mol) | RMSD (Å) | Interacting residues |
|---|---|---|---|---|---|
| 4a | -9.96 | 10.24 | -7.26 | 18.03 | LYS 53, LEU 171, |
| VAL 38, ALA 51, | |||||
| ASN 155, LEU 167, | |||||
| PHE 169 | |||||
| 4b | -10.02 | 34.14 | -7.96 | 14.21 | ASP 150, ASP 168, |
| LEU 74, TYR 323, | |||||
| PHE 169, ILE 146, | |||||
| ARG 149 | |||||
| 4c | -10.26 | 69.59 | -8.7 | 22.44 | GLY 110, ASP 112, |
| LEU 171, LEU 167, | |||||
| ALA 51, ALA 157, | |||||
| VAL 38, HIS 107 | |||||
| 4d | -10.48 | 56.76 | -8.98 | 19.18 | GLU 380, TRP 388, |
| ILE 164, GLN 283, | |||||
| ASN 288, ASN 317, | |||||
| PRO 385 | |||||
| 4e | -9.83 | 68.22 | -7.04 | 23.87 | TYR 35, HIS 148, |
| GLN 325, GLU 328, | |||||
| LEU 74, ARG 149, | |||||
| ASP 168, ASP 150 | |||||
Table 3: The binding energies, estimated inhibition constant, intermolecular energies and reference RMSD produced from docking for all studied compounds.
Molecular docking shown that the most interacting residues in the 4a compound active site included Lys 53, Leu 171, Val 38, Ala 51, Asn 155, Leu 167, Phe 169. 4b compound showed interaction with Asp 150, Asp 168, Leu 74, Tyr323, Phe 169, Ile 146, Arg 149. 4c compound was seen to interact with Gly 110, Asp 112, Leu 171, Leu 167, Ala 51, Ala 157, Val 38, His 107 and compound 4d interacted with Glu 380, Trp 388, Ile164, Gln 283, Asn 288, Asn317, Pro385 while compound 4e interacted with Tyr 35, His 148, Gln 325, Glu 328, Leu 74, Arg 149, Asp 168, Asp 150 as shown in Table 3.
Admet
In the field of drug discovery, due to antagonistic side effects and poor drug like nature, many potential drugs have failed during thier clinical trials. In this study, all newly synthesized compounds were subjected to ADMET studies to estimate the toxicity risk and drug-related properties in order to evaluate the drug-likeliness of these compounds. The drug likeliness properties of the studied compounds that estimated by ADMET were listed in Table 4 and the bi-plot is showed in Figure 3. The pharmacokinetic characteristics of the compound under study were established through six precalculated ADMET models provided by Discovery studio. Consider using a model with AlogP98 and PSA 2D descriptors and a bi-plot model with 95% and 99% confidence ellipses to accurately predict the cell permeability of the study compound. The log P used to calculate the lipophilicity, its behavior influences in a range of biological membranes which gives information about the ability of a compound to dissolve into lipophilic (non-aqueous). All newly studied compounds have good predictions of metabolic adsorption. In the toxicity assessment, all compounds except compounds 4a and 4b showed CYP2D6 inhibition and hepatotoxicity, indicating that these compounds are not toxic to the liver. BBB (Blood Brain Barrier) penetration assessment showed that compounds 4a and 4b compounds have undefined penetration, but other compounds (4c-4e) have moderate penetration. Therefore, compounds 4c, 4d and 4e may be suitable for central nerve system therapy.
| Compounds | aAbsorption | bBBB | cCYP2D6 | dHepatotoxicity |
|---|---|---|---|---|
| 4a | 0 | 3 | 0 | 0.179752 |
| 4b | 0 | 3 | 0 | 0.109022 |
| 4c | 0 | 2 | 1 | 0.497079 |
| 4d | 0 | 2 | 1 | 0.678977 |
| 4e | 0 | 2 | 1 | 0.510416 |
Note: aAbsorption: good absorption=0; moderate absorption=1; low absorption=2; bBBB level (Blood Brain Barrier): very high penetration=0; high penetration=1; medium penetration=2; low penetration=3; undefined penetration=4. cCYP2D6: non-inhibitor=0, inhibitor=1. dHepatotoxicity: noninhibitor=0, inhibitor=1.
Table 4: ADMET prediction of the optimized studied compounds.

Figure 3: Plot of Polar Surface Area (PSA) vs. LogP for a standard and test set showing the 95% and 99%
confidence limit ellipses corresponding to the Blood Brain Barrier and Intestinal absorption models. Note:
Computational study
In this study, we present results of a detailed investigation of the structural characterization of 4a, 4b, 4c and 4d using quantum chemical methods calculated by using the DFT (B3LYP) method with 6-31+G(D,P) basis set. The aim of this work was to explore the molecular dynamics and the structural parameters that govern the chemical behavior, and to compare predictions made from theory with experimental observations. In compounds the bond angles are listed in Table 5 and theoretical optimized in Figure 4.
| Bond angles (°) | B3LYP/6-31+g (d, p) 4a | Bond angles (°) | B3LYP/6-31+g (d, p) 4b |
|---|---|---|---|
| C14 _S20_N12 | 113.23 | C14_S20_N12 | 106.54 |
| S20_N12_C11 | 128.58 | S20_N12_C11 | 126.35 |
| S13_C11_N10 | 126.52 | S13_C11_N10 | 127.3 |
| N10_N9_C8 | 174.26 | N10_N9_C8 | 120.28 |
| S13_C11_N12 | 127.39 | S13_C11_N12 | 120 |
| N129O22_S20 | 130.1 | O22_S20_N12 | 106.24 |
| O21_S20_N12 | 61.7 | O21_S20_O12 | 122.07 |
| Bond angles (°) | B3LYP/6-31+g (d,p) 4c | Bond angles (°) | B3LYP/6-31+g(d,p) 4d |
| C23_O22_C3 | 118.72 | C13_C3_C6 | 119.56 |
| C7_N8_N9 | 121.03 | N8_N9_C10 | 116.54 |
| N9_C10 _N11 | 128 | S12_C10_ N9 | 111.37 |
| S12_C10_N9 | 111.15 | S12_C10_N11 | 120.93 |
| S12_C10_N11 | 120.8 | O21_S20_O22 | 104.04 |
| O20_S19_O21 | 104.14 | N11_C10_N9 | 127.69 |
| Bond angles (°) | B3LYP/6-31+g (d,p) 4e | ||
| C12_N19_C20 | 118.77 | ||
| C7_N8_N9 | 119.96 | ||
| N9_C10_N11 | 112.46 | ||
| S12_C10_N11 | 126.88 | ||
| O21_S22_O23 | 121.19 | ||
| S22_N11_C10 | 126.44 |
Table 5: Selected bond angles (◦), and theoretical calculations for the compounds.

Figure 4: Structure of theoretical optimized for compounds.
Analysis for forintier molecular orbitals
For physicists and chemists, molecular orbitals and their properties, such as energy, are extremely useful. In π-electron systems, their frontier electron density is used to predict the most reactive position, and in a conjugated system, he explained many forms of reactions [23]. Moreover, Frontier Molecular Orbitals (FMOs) are important orbitals for a molecule since they consist of two forms of HOMO (donating electron) and LUMO (accepting electron). These orbitals were able to depict the way a biomolecule binds to a receptor. The FMOs gap was huge. Characterized by the molecule's chemical reactivity and kinetic stability. The HOMO-LUMO energy gaps for the two compounds were calculated by 6-31+g (d, p) From the HOMO-LUMO orbital picture (Figure 5) and Table 6 it is found that the filled π-orbital (HOMO) in 4a and 4b is mostly located onthe nitrogyen atom while the unfilled antiπ-orbital (LUMO) is on benzene ring. When electron transitions take place, electrons are mainly transferred from nitrogen atom to the benzene ring.
| Compound | E (HOMO) | E (LUMO) | DE (LUMO-HOMO) |
|---|---|---|---|
| 4a | -0.23635 | -0.1907 | 0.16729 |
| 4b | -0.222 | -0.08 | 0.16175 |
| 4c | -0.23091 | -0.06362 | 0.17722 |
| 4d | -0.24084 | -0.07909 | 0.16175 |
| 4e | -0.20715 | -0.06429 | 0.14286 |
Table 6: Frontier orbital energy.

Figure 5: Frontier molecular orbital of the compounds.
Molecular Electrostatic Potential
The electronic density is related to the Molecular Electrostatic Potential (MEP), which is a useful descriptor in understanding electrophilic attack and nucleophile reactions, as well as hydrogen bonding interactions. Since it is through their potentials that the two organisms first "see" each other, the electrostatic potential V(r) is also well adapted for studying processes dependent on the "recognition" of one molecule by another, such as drug-receptor and enzyme-substrate interactions different. V(r)s can be calculated experimentally by diffraction or by statistical methods since it is a real physical property. Graphic models, especially MEP, have been used as a tool in conformational analysis by any researchers. The research of non-covalent interactions, primarily by investigating the electronic distribution in the molecule, is the study's key application. As a result, this approach was applied to determine the electronic distribution across the molecular surface for the component. MEP was measured at the 6-31+g(d, p) basis optimized geometry to visually consider the most likely sites of the molecules for an interaction with electrophilic and nucleophile species. While electrophilic reactivates are depicted in red, indicating the molecule's negative regions, nucleophile reactivates are depicted in blue, indicating the molecule's positive regions as shown in Figure 6.The sulfur and oxygen atoms are surrounded by a greater negative charge surface, becoming these sites potentially more favorable for electrophilic attack. In the MEP 4a, the main negative center is H5 atom attacked at C(3) of benzene ring, 4b the main negative center is N(12), 4C, 4d and 4e the main negative center is nitrogen atom which should be responsible for the interaction with the active drug-receptor sites.

Figure 6: Molecular electrostatic potential map of the compounds.
In this study, a series of five new benzenesulfonamide deivatives were synthesized by the reaction of aldehydes thiosemicarbazones derivatives with benzene sulphonyl chloride to form benzylidene- N-(phenylsulfonyl)hydrazine-1-carbothioamide derivatives. The different spectral techniques are used to confirm the structure of the novel synthesized compounds. 2-(4-(dimethylamino) benzylidene)-N-(phenylsulfonyl)hydrazine-1-carbothioamide (4e) and 2-(4-methoxy benzylidene)-N-(phenylsulfonyl)hydrazine-1- carbothioamide (4c) are the most potent anticancer derivatives against MCF-7 breast carcinoma cell lines. They also have the most potent antioxidant activities. Meanwhile, The 2-benzylidene-N- (phenylsulfonyl)hydrazine-1-carbothioamide (4a) and 2-(4-chloro benzylidene)-N-(phenylsulfonyl)hydrazine-1-carbothioamide (4d) have the lowest antioxidant potentials. The binding energy for the non-bonding interactions between the ligand (studied compounds) and receptor (4PYP (pdb code: 4FA2)) are calculated by using molecular docking to study anti-breast cancer activity of the studied compounds against human breast cancer MCF- 7 cells. The estimated binding energies, inhibition constant, intermolecular energies and reference RMSD produced from docking for all studied compounds are reported. These values show that all studied compounds formed stable complexes with receptor with high binding affinity. ADMET studies were performed to compute the toxicity risks and drug-relevant properties of the studied compounds. It is further noted from ADMET analysis that compounds 4c, 4d and 4e have good absorption, less toxic in the human liver and medium BBB penetration. Hence, these studied compounds (4c-4e) may therefore suggest as potential compounds against human breast cancer MCF-7 cells.
Sayed A Ahmed, Hussein S Mohamed wrote the introduction and experimental parts and do the experimental work for synthesis of new organic compounds, Osama M Ahmed wrote experimental parts and do the experimental biological work and Zeinab wrote and did theoretical work also share in the interpretation of data. A many prepared figures, wrote the results and discussion part and also shared in the interpretation of data.
The authors declare no competing interests.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
Citation: Mohamed HS, Hamza ZS, Nagdy AM, Ahmed SA, Ahmed OM (2022) Synthesis, Characterization, Biological Activities and Molecular Docking of New Benzenesulfonamide Drugs. J Clin Toxicol. 12:507.
Received: 02-Mar-2022, Manuscript No. JCT-22-49883; Editor assigned: 08-Mar-2022, Pre QC No. JCT-22-49883 (PQ); Reviewed: 05-Apr-2022, QC No. JCT-22-49883; Revised: 25-Apr-2022, Manuscript No. JCT-22-49883 (R); Published: 05-May-2022 , DOI: DOI: 10.35248/2161-0495.22.12.507
Copyright: © 2022 Mohamed HS, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.