Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Review Article - (2015) Volume 4, Issue 1
The metabolism of sulfur (S) compounds concurs to the maintain of cell homeostasis and tissue integrity in the human body. Sulfur chemical species act in all cells as anti-oxidant/scavenging agents or regulators of membrane stability/excitability. At the same time, they also exert tissue-dependent functions behaving as protective molecules of the liver and cardiovascular system, as modulators of the immune response, gut activity and CNS neurotransmitter signaling. The involvement of S compounds in human complex, chronic, disabling diseases at multifaceted pathogenesis is actually under investigation: altered levels of S metabolites could be in fact bio-indicators of impaired oxidation state in the body and their unbalance could be risk factors for disease onset. By the present review, we will discuss data from the literature which unearth an altered S biochemistry in human complex illnesses, taking as an example highly invalidating neuropsychiatry and pain perception diseases as autism spectrum disorders (ASD), schizophrenia and fibromyalgia. As well, we will depict herein the utility at applying to this area of the clinical research high resolving -omics strategies in combination with methodological tools which specifically explore S metabolism in patients. The perspectives of these kind of analyses would be the adoption of more valuable, personalized therapeutics protocols and, possibly, an improved bio-monitoring of patients, also including their response to treatments.
<Keywords: Sulfur metabolism; Human complex disorders; Autism; Schizophrenia; Fibromyalgia
As shown in the first part of this review dedicated to sulfur (S) metabolism in humans, the regulation of the molecular networks involving this element and the metabolic fluxes of S-containing amino acids (Met, Cys, HCys, and Tau) are overall orchestrated by diet, hormones, specific substrates as well as energy metabolism, on the basis of cell/tissue demands, aging and gender-linked factors. Since each S and S-AA biotransformation in humans takes part to numerous, fundamental functions at the interplay between environmental changes and chemical homeostasis, variations of one of the genes coding for enzymes of these paths, in relation to the type of genetic variant, can affect different tissues and anatomical districts or contribute to the pathogenesis of different possible pathological conditions. For instance, the autosomal recessive inherited disorder of transsulfuration characterized by homocystinuria, hyperhomocytinemia and hypermethioninemia due to a genetic defect of cystathionine-β- synthase (CBS), can provoke severe disturbances at the level of the Central Nervous System (CNS), the eye function and vision and/or the musculoskeletal and cardiovascular systems [1]. The second section of this review dedicated to S metabolism in human health deals instead with the possible contribution of unbalanced S biotransformations to human complex disorders, at multi-factorial, in most cases unclear, etiology and pathogenesis. Among complex human diseases, we can find, for instance, type II diabetes, obesity, a variety of cancer types, cardiovascular, neurodegenerative and neuropsychiatry disorders, epilepsy, pain perception, musculoskeletal and autoimmune disorders, as well as the often overlapping irritable bowel and chronic fatigue syndromes. These disorders can rise prenatally or during the early development or being typical diseases of a particular lifespan period, as adolescence or middle age, for instance menopause for women. Thus, it is usually accepted that human complex disorders derive from the interaction of genetic variance and environmental, lifestyle and lifespan factors. The genetic architecture of complex disorders is characterized by the involvement of multiple genes and/or proteins, differently from single-gene inherited disturbances. Common gene polymorphisms or allelic variants (single nucleotide polymorphisms, SNPs) of more than one gene have been found to concur, as vulnerability factors, to the development of a human complex disease [2,3]. In this last case, it is also possible that allelic variants of a same pleiotropic gene are linked to a number of different, but overlying, disorders, syndromes and altered responses [3,4]. Each gene variant and vulnerability factor involved can subtly and differentially shape phenotypes and traits in complex diseases [2,3]. Beside genetic variance, changed gene/protein expression patterns, epigenetic and metabolic variations in patients vs. controls can underlie state/trait factors of a complex disease [5]. Prevention and treatment of all these chronic and highly invalidating disturbances, at high costs for the whole community even in developing countries, represent main targets in clinical research. We therefore aim at presenting herein the “state-of-the-art” of the actually available data on the reported genetic, epigenetic and metabolic variations concerning S chemistry in patients affected by some among these complex diseases and in particular: the developmental brain autism spectrum disorders (ASD), the high invalidating psychiatric disease schizophrenia and the chronic pain syndrome fibromyalgia. We discuss these results trying to define the possible future perspectives and the methodological approaches to follow in the field.
Autism spectrum disorders
Autism spectrum disorders (ASD), encompassing Autistic Disorder, Asperger's Disorder, and Pervasive Developmental Disorder Not Otherwise Specified (PDDNOS), are characterized by relevant disturbances of the CNS function as impaired social behavior, deficits in emotion perception and non-verbal communication, accompanied by abnormal memory performance as well as disrupted cognitive and learning abilities. Alterations of brain development and CNS function in ASD occur prenatally or during the early childhood [6]. Despite the remarkable number of investigations on ASD, their etiology remains unclear. Genetics plays a major role, as revealed by the high (about 70%) concordance reported in twin studies and other investigations [6-8]. However, accordingly to the high symptoms’ variance, DNA investigations have also revealed a strong genetic heterogeneity in ASD, a feature which has contributed to consider them as non uniform genetic disturbances of brain development [9,10]. If common allelic variants have been found prevalent, combination of a rare and common variations or exclusively rare cannot be excluded in ASD, suggesting that gene variants and different protein functionalities underlie clusters of symptoms and dysfunctions. Next to molecular biology investigations, epigenetic and environmental factors of ASD are also receiving the highest consideration: the most currently accepted hypothesis among clinical researchers and neurobiologists is that ASD arise at the interface between vulnerability genes, epigenetic and environmental factors: changes in DNA/histone methylation patterns and altered gene expression are supposed to underlie ASD in the context of genetic vulnerabilities and particular lifestyles [11,12]. The study of S metabolism is part of this research field. Due to its involvement in methylation processes and gene expression, S metabolism has been investigated in wide cohorts of ill children and age-matched controls. Interestingly, altered transmethylation/transsulfuration metabolites together a 50% lower SAM/SAH ratio have been found in serum of autistic children vs. controls, suggesting an unbalanced Met metabolism accompanied by hypomethylation in ASD [13,14]. These studies are also supported by findings of an increased frequency of polymorphisms of Met re-methylation genes in ASD children than controls, such as the MethylenTetraHydroFolateReductase (MTHFR) 677C>T one which reduces the activity of this enzyme [15]. Other studies have reported lower circulating levels of GSH, Met and Cys, together increased levels of GSSG, the oxidized form of glutathione, in ill children [16-19], suggesting an impaired oxidative stress in autism disorders. The reduced ratio GSH:GSSG in ASD patients is a finding replicated in many studies, in blood or post-mortem brain using different technologies as HPLC, gas chromatography and others [16,20]. Moreover, impaired methylation capacity and altered circulating levels of HCys have been observed in autistic children and in their parents [21]. Either higher or lower HCys levels have been found in ASD or a different involvement of folate metabolism genes, implying the need to further investigate Met metabolism in these disorders and the possible presence of family clusters [15,21-24]. The immunological unbalance as well as dysbiosis and gut malfunction observed in autism [25,26] can derive from the reported changes in Met pathways. Mytochondrial defects have been also observed in ASD, further sustaining the role of oxidative metabolism in their pathogenetic mechanisms, accompanied by an impairment of pro-oxidant/anti-oxidant activities [27]. In addition, an impaired sulfotransferase (ST) detoxification capacity has been reported in ASD and genetic variants of ST Isoforms, the SULT1a, have been associated with autism [28]. Impaired sulfoconjugation and, as a consequence, an altered catecholamine catabolism, including cathecol-O-methyl-transferase (COMT) variants with altered activity, can affect noradrenergic and 5-HT cross-talks. It is worth noting that 5-HT levels have been found consistently increased in platelets of about 40% children with ASD, another among the most replicated biochemical features in biological studies concerning these disorders [29]. Hyperserotoninemia in autism can be explained by a polymorphism in the promoter region of the gene of the 5-HT transporter, at least in family clusters [30], but concurrent events are not avoided. Altered sulfation in autism can be even enhanced by the increased SO4 2- excretion observed in children with a diagnosis of autism, a finding related to the increased oxidative stress or to genetics features [31]. Sulfation seems in fact to play a main role during fetal development [32]. As well, some plasma AAs have been found altered in children with ASD [33,34].
The ASD neurochemistry is thus defined by heterogeneous genetic and epigenetic vulnerabilities: these can result in platelet hyperserotoninemia, impaired Met and folate metabolism, DNA hypomethylation and, possibly, gene expression up-regulation, increased oxidative stress and altered AA plasma profile; these dysfunctions can be present at different degrees, defining clusters of symptoms and phenotypes and the severity of the illness.
Schizophrenia and psychosis
Schizophrenia, a devastating behavioral disease, is characterized by delusions, thought disorder, hallucinations, psychosis and cognitive deficits. Schizophrenia affects the most basic human processes of perception, emotion and judgment at different degrees of severity [35]. As for autism, genetics studies of schizophrenia have shown heterogeneous and complex profiles, suggesting that the disease could originate from common and rare variants, but also from epigenetics alterations [36]. Beside the dopaminergic/serotonin hypothesis, other biochemical substrates are supposed to underlie schizophrenia and psychosis. As regards the topic of this review, an old story relates S-AAs, Met biochemistry and schizophrenia: in the early ‘60s, some authors observed that administration of Met together monoaminooxydase inhibitors (MAOI) worsened symptoms in schizophrenic patients [37,38]. Since that time, after a long period of disregard, a renovated interest is now emerging on Met pathways in neuropsychiatric disorders, depression, delusion and negative symptoms of psychoticrelated disorders and schizophrenia. First, genetic studies have involved genes of S biochemistry as vulnerability factors of the disease and a genetic hypothesis underlying psychosis and altered HCys metabolism has been also formulated [39]. Differently from autism, schizophrenic and bipolar psychotic patients characterized by Met metabolism dysfunction, consistently show elevated HCys plasma levels [40,41]. Moreover, as a risk factor to develop the disease, changes and variants of the 1 C cycle enzymes have been reported: in particular, as for ASD, the HCys remethylation enzyme MTHFR 677C>T has been linked to psychotic behavior [42-44]. Other S-related genes have been implicated in schizophrenia, as DNA variation of SULT4a1, a sulfotransferase isoform expressed in the brain only which specifically promotes sulfation of catecholamine [45], or Met sulfone reductase [46]. Plasma levels studies have linked variation of plasma S-AAs as Met and Cys to different phases of this invalidating mental illness: for instance, Met was found lower in psychotic patients unresponsive to atypical antipsychotic drugs, whereas high Met was reported in drugfree patients with schizophrenia. Others have shown low S-AAs levels in psychosis. Finally, some authors have reported that an altered plasma Tau/Met-Ser ratio can be a powerful biomarker of acute psychosis [47]. Investigations on platelet STs in patients with mood disorders have shown an increased enzyme activity in bipolar disorder [48]. These data, albeit in part discordant, suggest an imbalance of S metabolism in psychosis and schizophrenia. As aforementioned, a number of studies have reported increased circulating levels of HCys in schizophrenic and bipolar patients [49,50]. High HCys in schizophrenia has been related to the above reported genetic variations, but several new findings are also in support of epigenetic causes. The amount of HCys in tissues and blood depends from Met metabolism balance and, in particular, from the relative activities of Met transmethylation and remethylation enzymes, regulated by the intake of folic acid and B group vitamins. High circulating levels of HCys have been related not only to neuropsychiatric disorders but also to cardiovascular diseases, diabetes and neurodegenerative diseases, indicating that its metabolism exert pleiotropic effects in the body. An interesting finding has shown the significant reduction of transthyretin, a protein transporter of the circulating thyroid hormone T4 and retinol, in psychosis [51]. Since HCys reduces the active form of transthyretin [52], cognitive impairment in psychosis and schizophrenia could be linked to high HCys levels and low transthyretin. The hypothesis formulated by Costa and coauthors starts instead from the early Met studies on schizophrenia and relates these findings to an hypermethylation of specific genes, provoking their down-regulation, in schizophrenic patients [53,54]. The susceptibility seems linked to gender variables [54]. Interestingly, Met metabolism and dopamine transmission have been found interlaced in schizophrenia, a finding needing replication [55]. Thus, schizophrenia and psychosis are characterized by heterogeneous genetics and, at a different degree, by dopamine/ monoamine imbalance, altered sulfation, S metabolism changes and, possibly, hypermethylation, gene expression down-regulation patterns and transthyretin deficit. Mytochondrial dysfunctions are also emerging [56].
Fibromyalgia
The metabolism of S, Met and other S-AAs has been extensively investigated in rheumatic diseases and osteoarthritis. Herein, we will call rather attention on unspecified muscle pain disorders as fibromyalgia. This syndrome is characterized by a constellation of pain symptoms and overlap with neuropsychiatric and gastrointestinal diseases [57]. Despite being one of the most frequent diagnoses in clinical rheumatology practice, fibromyalgia etiology and pathogenesis remain elusive. Fibromyalgia syndrome has been related to disturbances of the hypothalamic–pituitary axis and neurotransmission defects, involving excitatory amino acids, catecholamines, substance P and 5-HT [58- 60]: patient's symptoms may derive from poor stressor modulation, sensitization of specific nociceptor neurons and pain threshold diminution in response to multiple environmental factors, such as mechanical or emotional trauma, chronic stress or even infections. In substance, all patients with fibromyalgia, in high prevalence women, report a diminished pain perception threshold together a greater vulnerability to diverse stressors. Concerning the role of S metabolism, its indirect involvement in this syndrome is supported by the clinical efficacy of oral administrations of SAM (Samyr) reported in patients with fibromyalgia [61]. This has been mainly ascribed to the fact that SAM is a methyl donor in epinephrine or melatonin formation, two molecules involved in sleep-arousal regulation and mood/anxiety tonus [62], frequently altered in patients with fibromyalgia. Despite treatment with SAM is effective in fibromyalgia, relatively few studies have been conducted on S metabolism in this field of medical pathology. Significantly lower levels of ATP, a trend toward higher Ca2+ and Mg2+ content in platelets as well as markedly reduced plasma levels of S-AAs Met and Tau, together low phenylalanine and tyrosine, the precursor of catecholamines and thyroid hormones, have been obtained in patients with fibromyalgia vs. healthy control subjects [63-65]. Intestine malabsorption could be a cause of the observed reduction of plasma AAs in patients, this accompanied by gut microbiome alteration and disbiosis [66]. Epigenetic alteration and changes in methylation patterns are also emerging in fibromyalgia [67].
These results, albeit preliminary, are in support of an imbalance of S, catecholamine and purinergic metabolism in patients with fibromyalgia. These results can be ascribed to a concomitant increased oxidative stress in patients with fibromyalgia, as supported by some authors: in fact, in other studies,lower serum levels of catalase and GSH have been found in serum of fibromyalgic subjects [68].
Sulfur, diet and human complex diseases
Nutritional aspects cannot be underestimated when investigating S metabolism in ASD, schizophrenia and fibromyalgia, where a genetic and epigenetic impact has been reported. Diet and biological research on these three complex diseases are the two faces of a same medal: on one side, the nutritional profile of patients should be monitored since diet can alter results on the search of metabolic disturbances in patients, especially those regarding plasma/cell levels of S-compounds or oxidative stress biomarkers in patients. On the other side, the appraise of patients’ nutritional state is part of the disease itself, since stressors or other triggering factors can influence feeding behavior in vulnerable subjects, provoking nutritional and metabolic deficits. In some individuals, vulnerable genes are even directly implicated in the metabolic processes altered by an unbalanced diet, enhancing therefore some symptom features rather than others.
A normal, equilibrated diet, along with a good supply of proteins, gives the required amount of S. However, the complexity of S metabolism and its regulation makes difficult to define its real requirement for health care in different lifespan stages and pathological conditions [69]. Moreover, Met metabolism is dependent of vitamins and essential cofactors: thus, it is evident that diet can strongly influence S-compounds’ metabolism in the body [70,71]. Stressors, low stress coping, low-quality lifestyle, drug abuse, alcoholism and stress-related pathologies potentially lead to incorrect alimentary choices/habits and even taste changes [72]. Unbalanced diet can affect methylation patterns and epigenomics [73,74]. The recommended daily allowance (RDA) suggests to ingest at least 13 mg Kg-1 body weight per day of S, but other sources contrast these values and recommend daily doses ≥ 20 mg Kg-1 body weight. Some authors have reported that the different stages of the life span require variable S assumption from diet [75]. On the other side, some authors have reported that Met diet restriction increases GSH intra-cell levels and lifespan in mouse strains through feed-back adaptation mechanisms [76]. Therefore, additional investigation would provide an improved understanding of factors determining S and S-AAs bioavailability in healthy subjects and patients.
Sulfur metabolism and –Omics techniques in ASDs, schizophrenia and fibromyalgia
The study of S-AA metabolism is carried out through specific isotope tracers taken by S compounds [77]. Furthermore, the development of high-resolving techniques as HPLC coupled to UV or electrochemical and fluorescence detection has improved the study of S metabolites in body fluids or cells [78]. A valuable, specific and sensitive measurement of S-AAs, GSH/GSSG ratio, SAM/SAH ratio can in fact provide useful information on cell redox state, DNA methylation and Met metabolism. On the other side, a multi-factorial approach is thus the most suitable, implying the identification of clusters and groups of patients within a pathological condition, presumably showing distinct biochemistry patterns, symptoms or responses to treatment. Database can be evaluated through suitable multivariate statistical analysis, including cluster and principal component analysis. Recognizing the existence of biochemical clusters within schizophrenia and fibromyalgia could further support the notion that these disorders are not “single”, “fixed” pathological entities but rather spectrum disorders.
The new emerging -omics technologies are, by now, the best approach to investigate biological correlates of complex, chronic and invalidating disorders as ASDs, schizophrenia and fibromyalgia, permitting to monitor the numerous disease’s variables. These techniques offers a “full screen” vision of patients’ biology, consisting in DNA array genomic/transcriptomic/methylomic analyses, in the proteomic evaluation of all expressed proteins as well as the measure of metabolites, substrates (metabolomics) and drug levels in cells, tissues or body fluids of patients during the different phases of the disease and pharmacological therapy. Proteomics and metabolomics apply LC/MSMS techniques which enable the measurement of numerous proteins and substrates. Hyphenated HP-LC techniques are also improving resolution in the field [79].
If a main obstacle to these investigations consists in the highcosts, this can be circumvented by the participation into multicenter laboratory studies. The-omics tool permits to simultaneously investigate multiple, potentially involved molecular mechanisms and systems in cells and tissues, from DNA to metabolic substrates. As an example, Figure 1 is a schematic representation of those molecular substrates and factors potentially underlying ASD, schizophrenia and fibromyalgia together the presumed triggering genetic, epigenetic and environmental variables.
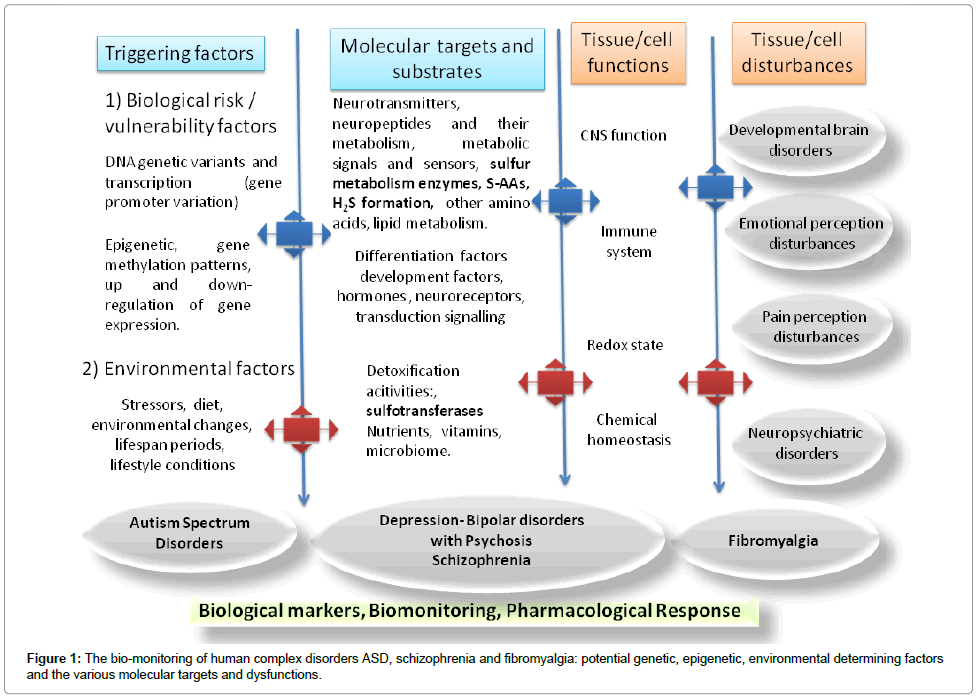
Figure 1: The bio-monitoring of human complex disorders ASD, schizophrenia and fibromyalgia: potential genetic, epigenetic, environmental determining factors and the various molecular targets and dysfunctions.
For -omics investigations, the analysis of body fluids as blood, serum, plasma, saliva or peripheral cell models as circulating lymphomonocytes, platelets or erythrocytes and tissue autopsy, can be integrated with the traditional protein specific assays and/or postmortem/ animal studies which apply to the search of a single or few specific biomarkers. In the case of S metabolism, specific evaluation consists, as indicated before, in the search of changes of Met metabolites as HCys in body fluids. Applying -omics techniques would permit to characterize specific S metabolic profiles also targeting disease’s genetic/epigenetic, redox unbalance in human disorders as well as pharmacological treatment by drug metabolism evaluation and patient response to treatments. This approach endows with a robust, powerful tool of investigation of complex disorders as ASD, schizophrenia and fibromyalgia. New relationships between metabolic paths and systems could be found, providing signatures, maps and pathophysiological networks. In autism research, proteomics and metabolomics are leading towards new diagnostic and therapeutic perspectives [80- 82]. The same is occurring for schizophrenia [83,84] or fibromyalgia [85,86]. Application of –omics strategies in these diseases have also involved the therapeutic monitoring of pharmacological response [87], nutritional aspects and microbioma evaluation [88].
The targeting of S biology represents a main issue in human pathology which could be better defined through a multidisciplinary approach, encompassing molecular biology, biochemical, pharmacological, nutritional and statistical evaluations. The -omics tool can be a winning strategy to monitor impaired metabolic redox states, networks and patterns in complex, chronic and invalidating human diseases as ASD, schizophrenia and fibromyalgia, even coupled to specific S metabolism evaluations. This would permit to know more about their pathogenesis, possibly permitting to apply personalized therapies. The understanding of redox/ROS modulation mechanisms in cells and tissues and their manipulation would represent a main goal in molecular pathology and treatment of a variety of human diseases. The role of transsulfuration enzymes, their regulation and H2S formation should be further evaluated as well as metallothioneins and mitochondrial proteins. This approach could help to understand the almost unclear etiology of these diseases, as well as to detect clusters of symptoms and biochemical changes in these disorders, improving pharmacological investigations.