Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2015) Volume 4, Issue 3
Alzheimer's disease (AD) is characterized by several pathologies, as this disease involves neuropathological lesions in the brain. Indeed, a wealth of evidence suggests that β-amyloid is central to the pathophysiology of AD and is likely to play an early role in this intractable neurodegenerative disorder. AD is the most prevalent form of dementia, and current indications show that twenty-nine million people live with AD worldwide, a figure expected to rise exponentially over the coming decades. Clearly, blocking disease progression or, in the best-case scenario, preventing AD altogether would be of benefit in both social and economic terms. However, current AD therapies are merely palliative and only temporarily slow cognitive decline, and treatments that address the underlying pathologic mechanisms of AD are completely lacking. While familial AD (FAD) is caused by autosomal dominant mutations in either amyloid precursor protein (APP) or the presenilin (PS1, PS2) genes. First, we have reviewed 2D QSAR, 3D QSAR, CoMFA, CoMSIA and docking for GSK-3α and GSK-3β with different compounds to find out their structural requirements. Next, we develop a QSAR for GSK-3β, because is one of the most important enzymes that intervenes in neuropathological disease such as Alzheimer. QSAR could play an important role in studying these GSK-3 inhibitors. For this reason we developed QSAR models for GSK- 3β, LDA, ANNs and CT from more than 40000 cases with more than 2400 different molecules inhibitors of GSK-3β obtained from ChEMBL database server; in total we used more than 45000 different molecules to develop the QSAR models. We used 237 molecular descriptors calculated with DRAGON software. The model correctly classified 1310 out of 1643 active compounds (79.7%) and 24823 out of 26156 non-active compounds (94.9%) in the training series. The overall training performance was 94.0%. Validation of the model was carried out using an external predicting series. In this series the model classified correctly 757 out of 940 (80.5%) active compounds and 14 166 out of 14 937 non-active compounds (94.8%). The overall predictability performance was 94.0%. In this work, we propose five types of non Linear ANN and we show that it is another alternative model to the already existing ones in the literature, such as LDA. The best model obtained was RBF 166:166-402-1:1 which had an overall training performance of 94.2%. All this can help to design new inhibitors of GSK-3β. The present work reports the attempts to calculate within a unified framework probabilities of GSK-3β inhibitors against different molecules found in the literature.
<Keywords: GSK-3β; QSAR; Artificial neural network; Linear neural network; Linear discriminant analysis
Glycogen synthase kinase-3 (GSK-3) has two isoforms, GSK-3α and GSK-3β, [1] and they are serine/threonine kinases involved in numerous cellular processes and diverse diseases as Alzheimer disease, cancer, and diabetes. GSK-3α and GSK-3β have been shown to be present in mammals and the latter is specifically expressed in the central nervous system [2,3]. In particular, GSK-3β is well known to play critical roles in oxidative stress-induced neurodegenerative diseases such as Alzheimer´s disease (AD) [2,4]. Despite intensive investigation into the physiological roles of GSK-3 isoforms, the basis for their differential activities remains unresolved. A more comprehensive understanding of the mechanistic basis for GSK-3 isoform-specific functions could lead to the development of isoform-specific inhibitors [5]. GSK-3β knock-out mice die in utero [6], whereas GSK-3α knockout mice are viable and display improved glucose tolerance in response to glucose load and elevated hepatic glycogen storage and insulin sensitivity [7,8].
Alzheimer´s disease [9] is a serious and degenerative disorder that causes a gradual loss of neurons, and in spite of the efforts realized by the big pharmaceutical companies of the world, the origin of this pathology is still not very clear. β-amyloid (Aβ) is an important protein implicated in the pathogenesis of AD, but the mechanism by which it causes neurotoxicity is still unknown [10,11]. In particular, there are few literature reports to study the direct link between the pathological hyperphosphorylation of tau protein, a microtubule associated protein, and the formation of neurofibrillary tangles (NFT) [12]. The last decades had marked a very significant era of AD research. During this period, the nature of amyloid plaques and NTFs, the two histopathological hallmarks of AD, had been elucidated. Recent research efforts have led to several hypotheses to explain AD. Amyloid β toxicity is believed to play a primary role in the development of AD [13]. GSK-3β activity may increase with aging [14], which is consistent with the fact that aging is the most important risk factor for AD. Both in vitro and in vivo studies have demonstrated that inhibition of GSK- 3β, can reverse hyperphosphorylation of tau and prevent behavioral impairments in mice [15-20]. These studies make GSK-3β inhibition very attractive as a therapeutic target for AD [21].
In the last years, a number of publications have been published suggesting GSK-3 as a target for the treatment of AD. There are two isoforms of GSK-3, GSK-3α and GSK-3β, both sharing a high homology at their catalytic site, but the α form possesses an extended N-terminus with respect to the β form [22,23]. The phosphorylation of proteins by GSK-3 is an important link in neural function [24-26]. There are two characteristic neuropathological hallmarks of AD, Neurofibrillary Tangles (NFT’s) and an increased production of amyloid beta (Aβ) peptides, where NFT’s are composed of highly phosphorylated forms of the microtubule-associated protein tau [27] and studies have shown that GSK-3 is one of the main in vivo players of phosphorylation of tau protein [28]. It has been reported that Lithium, a GSK-3 inhibitor, blocks production of Aβ peptides by interfering with APP cleavage at γ-secretase step, where the target for Lithium is GSK-3α [22,29]. Phiel et al. [29] showed that selective reduction in concentration of the α isoform led to a decrease in the concentration of Aβ40 and Aβ42, primary constituents of amyloid plaques in AD. Thus, inhibition of GSK-3α could potentially provide dual therapy against AD, preventing the buildup of amyloid plaques and of neurofibrillary tangles [29-31].
GSK-3β is a serine/threonine kinase and is thought to be a key factor for aberrant tau phosphorylation [32]. Activated GSK-3β coexists with progression of NFT’s and neurodegeneration in the AD brain [33- 35]. A conditional GSK-3β overexpressing transgenic mouse exhibits persistent tau hyperphosphorylation, pretangle-like somatodendritic localization of tau, neuronal death in hippocampus and cognitive deficits [36,37]. These studies suggest that GSK-3β is associated with AD progression, and GSK-3β inhibition is expected to be a promising therapeutic approach for AD.
In this sense, quantitative structure-activity relationships (QSAR) could play an important role in studying these β and γ-secretase inhibitors. QSAR models are necessary in order to guide the β and γ-secretase inhibitors.
On the other hand, QSAR models can be used to explore the relationships between the structural spaces of compounds as inhibitors for specific enzymes, such as MAO inhibitors [38], HIV-1 integrase inhibitors [39], and/or protease inhibitors [40] or tyrosinase inhibitors [41-43]. In fact, almost all QSAR techniques are based on the use of molecular descriptors, which are numerical series that codify useful chemical information and enable correlations between statistical and biological properties [44,45]. Recently, the field has moved from small molecules to proteins and other systems. For instance, González-Díaz et al. have discussed the use of these methods but only from the point of view of proteins [46]. Later, some groups have published different papers in one special issue on QSAR but they have been also restricted to the field of protein and proteomics [47-53]. In other recent issue, guestedited by González-Díaz [54] a series of papers have been published, devoted to QSAR/QSPR techniques for low-molecular-weight drugs [54-63]. Most recently, Prado-Prado et al. [64] have published a mt- QSAR for anti-parasitic drugs. This year we have published another issue [65] focused on QSAR/QSPR models and a graph theory used to approach Drug ADMET processes and Metabolomics [66-73]. Last, one of the most recent issues published has discussed the applications of QSAR in Pharmaceutical Design [74-83].
The functions of GSK-3 and its implication in various human diseases have triggered an active search for potent and selective GSK-3 inhibitors [12] in the last years. QSARs can be used as predictive tools for the development of molecules [84,85]. The QSAR approach involves the development of models that relate the structure of drugs with their biological activity against different targets [86,87]. Furthermore, there are multiple chemometric approaches that can, in principle, be selected for this step. Multiple linear regression (MLR), LDA, partial least squares (PLS) and different kinds of artificial neural networks can be used to relate molecular structure (represented by molecular descriptors) with biological properties. The ANNs are particularly useful in QSAR studies in which the linear models fit poorly due to high data complexity; an example was the work of Prado-Prado et al. in which four types of non-ANN were developed to calculate within an unified framework probabilities of antiparasitic action of drugs against different parasite species [64,88,89]. There are several different kinds of ANN and these include multilayer perceptron (MLP), radial basis functions (RBF) and PNNs; the latter ANN is a variant of RBF systems. In particular, PNN is a type of neural network that uses a kernel-based approximation to form an estimate of the probability density functions of classes in a classification problem [90]. In the present work, we have reviewed previous works based on 2D-QSAR, 3D-QSAR, CoMFA, CoMSIA and docking techniques, which studied different compounds to find out the structural requirements. Last, in this review, we developed quantitative structure-activity relationships (QSAR) models for GSK- 3β, linear discriminant analysis (LDA) [91] and linear artificial neural networks (ANNs) from more than 40000 cases with more than 24000 different inhibitors of GSK-3β obtained from ChEMBL database http:// www.ebi.ac.uk/chembldb/index.php/target/browser/classification [92,93]; in total we used more than 45000 different cases to develop the QSAR models. In addition, we did a study of different fragments that exist in the molecules of the database in order to see which fragments had more influence in the activity, and which fragments interact more with the protein. As there are very studies with GSK-3β that can be found in the literature the design of new inhibitors of this enzyme is very important for study of the neurodegenerative diseases [94,95]. The topics reviewed, discussed, and/or reported in this paper are:
1. Studies of GSK-3α inhibitors
1.1. 2D-QSAR for 3-anilino-4-phenylmaleimides
1.2. 3D-QSAR and docking of 3-anilino-4-phenylmaleimides
1.3. QSAR studies of Some GSK-3α Inhibitory pyrimidines
2. Studies of GSK-3β inhibitors
2.1. Design, synthesis and structure-activity relationships of 1,3,4-oxadiazole derivatives
2.2. Linear/Nonlinear Regression Methods for Prediction of Glycogen Synthase Kinase-3β Inhibitory Activities
2.3. Molecular modeling, docking and 3D-QSAR studies for maleimides
2.4. Molecular Docking and biological testing of new GSK-3β inhibitors
2.5. 3D-QSAR Modeling of Paullones
2.6. Modeling of Binding Mode of Benzo[e]isoindole-1,3-diones
3. QSAR studies of GSK-3β
3.1. Theoretical study of GSK-3β: Neural Networks QSAR studies
2D-QSAR for 3-anilino-4-phenylmaleimides
Sivaprakasam et al. [31] reported in their study a 2D-QSAR exploration of the physicochemical (hydrophobic, electronic, and steric) and structural requirements among 3-anilino-4-phenylmaleimides toward GSK-3α binding. Using Fujita-Ban and Hansch QSAR analyses, electronic and steric interactions at the 4-phenyl ring and hydrophobic interactions at the 3-anilino ring were shown to be crucial. Hanschtype QSAR was still widely used in the lead optimization stage of synthetic and other projects.
Fujita-Ban analysis of 3-anilino-4-phenylmaleimides revealed that certain structural features such as Cl, OCH3, and NO2 mono substitution at any position around the 4-phenyl ring were favorable for GSK-3α inhibition. Substituents at the 3-anilino ring such as 3-Cl, 4-Cl, 5-Cl, 3-COOH, 4-OH, and 4-SCH3 were positively and 3-OH was negatively correlated with GSK-3α inhibitory activity.
Through Hansch QSAR analyses, they found that the GSK-3α inhibitory activity was enhanced by: 1. Electron-withdrawing, bulky ortho substituents at 4-phenyl ring; 2. 4-chloro substitution around anilino ring; 3. 3-anilino rather than 3-N-methylanilino derivatives; 4. Hydrophobic meta substituents on the anilino ring. Overall, QSAR models 13a and 14a suggested electronic and steric effects at the 4-phenyl ring and hydrophobic effects at the 3-anilino or 3-N-methylanilino ring were crucial. Their 2D-model (Figure 1) illustrated these effects which are essential for binding the maleimides to the GSK-3α enzyme. Their analysis provided key information regarding ligand–target interactions which they believed would help medicinal chemists to design more potent GSK-3α inhibitors.
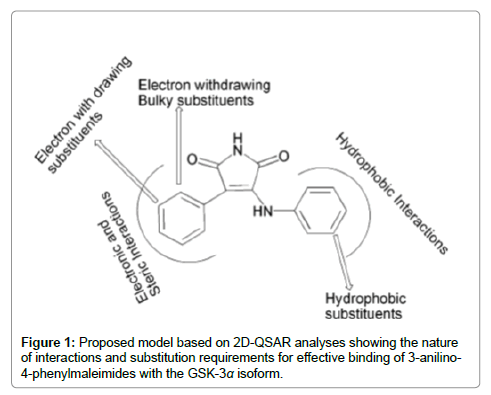
Figure 1: Proposed model based on 2D-QSAR analyses showing the nature of interactions and substitution requirements for effective binding of 3-anilino- 4-phenylmaleimides with the GSK-3α isoform.
3D-QSAR and docking of 3-anilino-4-phenylmaleimides
3D-QSAR analyses were reported in this article [96], using CoMFA and CoMSIA and molecular docking studies on 3-anilino-4- phenylmaleimides as GSK-3α inhibitors, in order to better understand the mechanism of action and structure-activity relationship of these compounds. The comparison of the active site residues of GSK-3α showed that all the key amino acids involved in polar interactions with the maleimides for the β isoform were the same in the α isoform, except for Asp133 in the β isoform, which was replaced by Glu196 in the α isoform. The authors prepared a homology model for GSK- 3α and showed that the change from Asp to Glu should not affect maleimide binding significantly. Their best CoMFA model contained steric and electrostatic fields and had n = 56, q2 = 0.844, r2 = 0.942, SEE = 0.104, F = 162.49 and r2 pred = 0.779 for five components. CoMFA electrostatic contours revealed that increased negative charge at the meta position of the 4-phenyl ring was favorable for the activity. They found that electron withdrawing groups at the meta and para positions around the anilino ring were important for enhancing activity.
Electron-withdrawing bulky ortho substituents on the 4-phenyl ring were conducive to GSK-3α inhibition. CoMSIA model showed the importance of hydrogen bond donor groups on these ligands for enhanced activity. The best CoMSIA model (S + E + D) had n = 56, q2 = 0.833, r2 = 0.932, SEE = 0.113, F = 111.67 and r2 pred = 0.803 for six components. Comparatively, 3-N-methylanilino derivatives were less active than 3-anilino derivatives.
Docking studies revealed the binding poses of three subclasses of these ligands, namely anilino, N-methylanilino and indoline derivatives, within the active site of the β isoform, and helped to explain the difference in their inhibitory activity.
QSAR studies of some GSK-3α inhibitory pyrimidines
Jamloky et al. studied in this paper [22] a series of pyrimidines which was performed to gain structural insight into the binding mode of the molecules to the GSK-3α. The molecular modeling studies were performed using CS Chem. Office 2001 molecular modeling software version 6.0. MOPAC module was used to minimize the energy and calculate the descriptors. The thermodynamic and steric features of the pyrimidines were highly correlated with GSK-3α inhibitory activity. The positive coefficient of PMI-Y in the model suggested that the presence of bulky substituents positioned towards the Y-axis of the molecule would enhance the GSK-3α inhibitory activity. The observation supports the hypothesis that the presence of the bulky substituents like bromine with inherent hydrophobic character may be involved in the nonspecific interaction with the ATP binding site. The results of the study suggested that the introduction of bulky groups at C-5 position of the hydrophobic interaction with the ATP binding site of the enzyme may be attributed to the strain exerted by the two adjacent phenyl rings on the planar pyrazolo (3,4-b) pyridine ring, thereby partly disrupting the hydrogen bonding interaction between nitrogen in the pyrazolo group and the complementary group in the enzyme.
Design, synthesis and structure-activity relationships of 1,3,4-oxadiazole derivatives
Saitoh et al. [97] reported design, synthesis and structure–activity relationships of a novel series of oxadiazole derivatives as GSK-3β inhibitors. Among these inhibitors, compound 20x showed highly selective and potent GSK-3β inhibitory activity in vitro and its binding mode was determined by obtaining the X-ray co-crystal structure of 20x (Figure 2) and GSK-3β (Figure 3). The hydrogen bonding interaction of the benzimidazole core with the hinge region and the oxadiazole with Asp200 were observed. Additionally, the interaction of 4-methoxyphenyl group with Arg141 was also observed.
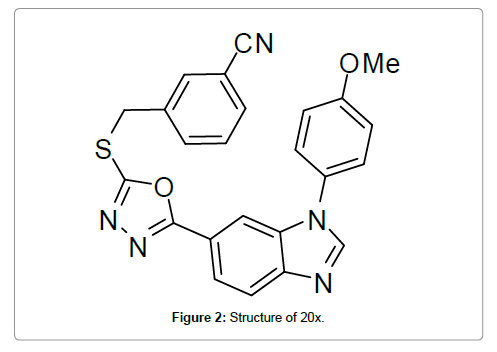
Figure 2: Structure of 20x.
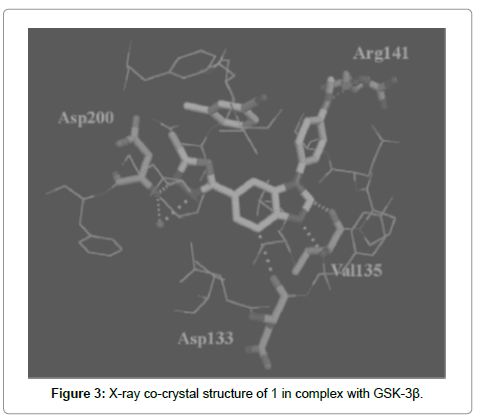
Figure 3: X-ray co-crystal structure of 1 in complex with GSK-3β.
Linear/nonlinear regression methods for prediction of glycogen synthase Kinase-3β inhibitory activities
Freitas et al. [98] applied linear/nonlinear regression methods as multiple linear regression (MLR), artificial neural network (ANN), and support vector machines (SVM) with a series of glycogen synthase kinase-3β (GSK-3β) inhibitors using calculated Dragon descriptors. Few variables were selected from a pool of calculated Dragon descriptors through three different feature selection methods, namely genetic algorithm (GA), successive projections algorithm (SPA), and fuzzy rough set ant colony optimization (fuzzy rough set ACO). The fuzzy rough set ACO/SVM-based model gave the best estimation/ prediction results, demonstrating the nonlinear nature of this analysis and suggesting fuzzy rough set ACO, introduced in chemistry for the first time, as an improved variable selection method in QSAR for the class of GSK-3β inhibitors. MLR yielded QSAR models only reasonably predictable, with r2 ranging from 0.77 to 0.81 and r2 test of 0.67 to 0.76, ANN and specially SVM were capable of estimating and predicting biological activities very accurately.
Molecular modeling, docking and 3D-QSAR studies for maleimides
Hwan-Kim et al. [99] carried out molecular modeling and docking studies with three-dimensional quantitative structure relationships (3D-QSAR) to determine the correct binding mode of glycogen synthase kinase 3β (GSK-3β) inhibitors. For the 3D-QSAR (CoMFA and CoMSIA), they used 51 substituted benzofuran-3-yl-(indol-3-yl) maleimides. Two binding modes of the inhibitors to the binding site of GSK-3β were analyzed. The binding mode 1 yielded better 3D-QSAR correlations using both CoMFA and CoMSIA methodologies. The three-component CoMFA model from the steric and electrostatic fields for the experimentally determined pIC50 values had the following statistics: R2(cv) = 0.386 and SE(cv) = 0.854 for the cross-validation, and R2 = 0.811 and SE = 0.474 for the fitted correlation. F (3.47) = 67.034, and probability of R2 = 0 (3.47) = 0.000. The binding mode suggested by the results of this study was consistent with the preliminary results of X-ray crystal structures of inhibitor-bound GSK-3β. The 3D-QSAR models were used for the estimation of the inhibitory potency of two additional compounds.
Molecular docking and biological testing of new GSK-3β inhibitors
Lavrovskii et al. [100] used a series of new heteroaryl-substituted oxadiazole-5-carboxamide inhibitors of GSK-3β. Molecular docking was used for the rational selection of synthesized compounds for the subsequent biological testing. It was established that the inhibitory activity of the synthesized compounds strongly depends on the character of substituents in the phenyl ring and the nature of terminal heterocyclic fragments. The most active compounds inhibit GSK-3β at IC50 in the micro molar range and could be considered as potential drug candidates.
3D-QSAR modeling of paullones
Osolodkin et al. [101] carried out a 3D-QSAR study which suggested ways of modification of the molecule to increase its physiological activity. A comparative molecular field analysis (CoMFA) [7] and a comparative molecular similarity indices analysis (CoMSIA) [8] are among the most widely used 3D-QSAR methods. The energy of Van der Waals and electrostatic interactions of a probe atom (with the charge +1) with molecules of the training set (CoMFA) or the electrostatic, Van der Waals, hydrophobic, and donor/acceptor similarity indices (CoMSIA) were used as descriptors. The equation for activity prediction was derived using the partial least squares (PLS) method. The advantages of the methods were the ability of graphic representation of PLS model coefficients and the fact that they allowed the user to suggest substitutions affecting activity and/or selectivity of the molecules. The authors built a new 3D-QSAR model for GSK-3β inhibition by paullones by means of the CoMFA method. This model can be used as a guide for designing new paullone GSK-3β inhibitors.
Modeling of binding mode of Benzo[e]isoindole-1,3-diones
Yang et al. [102] synthesized benzo[e]isoindole-1,3-dione derivatives and the effects on GSK-3β activity and zebrafish embryo growth were evaluated. A series of derivatives showed obvious inhibitory activity against GSK-3β. The most potent inhibitor, 7,8-dimethoxy-5-methylbenzo[e]isoindole-1,3-dione, showed nanomolar IC50 and obvious phenotype on zebrafish embryo growth associated with the inhibition of GSK-3β at low micro molar concentration. The interaction mode between this compound and GSK-3β was characterized by computational modeling. To rationalize the structure-activity relationships of these compounds, the binding modes of the most potent inhibitors 8a and 8b (Figure 4) were modeled using docking simulations. Compounds 8a and 8b were docked into the ATP binding site of GSK-3β, and the binding modes of the lowest energy were analyzed. Compounds 8a and 8b fit the ATP pocket of GSK-3β well. The maleimide motif of type II formed a pair of hydrogen bonds with the hinge region (Glu133 and Val135) of GSK-3β, similar to the binding mode of other known maleimides GSK-3β inhibitors. The two methoxy oxygen atoms formed another two hydrogen bonds with the positively charged Lys85. The methyl group of the methoxy at C-8 position docked to the small back cleft of GSK-3β. This binding mode explicitly explained the important role of the two methoxy groups at C-7 and C-8 positions. Another result was the 4-ethyl group of 8b docks to the minor hydrophobic pocket made up of Ile62 and Val70 in front of the ATP binding site of GSK-3β (Figure 5), which contributed to its higher binding affinity compared to 8a. The docking results also provided a template to understand the structure-activity relationships of other compounds.
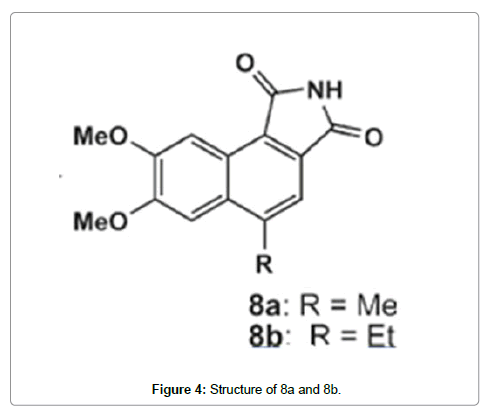
Figure 4: Structure of 8a and 8b.
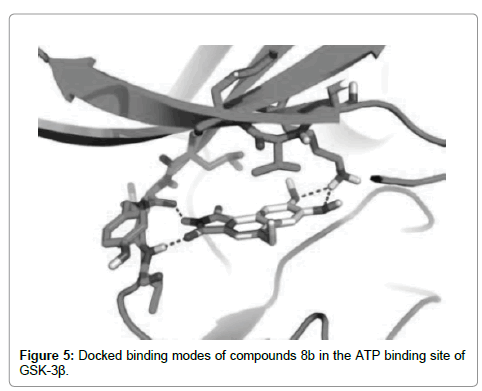
Figure 5: Docked binding modes of compounds 8b in the ATP binding site of GSK-3β.
Theoretical study of GSK-3β: Neural Networks QSAR studies for the design of new inhibitors using 2D-descriptors
Alzheimer´s disease [9] is a serious and degenerative disorder that causes a gradual loss of neurons, and in spite of the efforts realized by the big pharmaceutical companies of the world, the origin of this pathology is still not very clear. β-amyloid (Aβ) is an important protein implicated in the pathogenesis of AD, but the mechanism by which it causes neurotoxicity is still unknown [10,11]. In particular, there are few literatures report to study the direct link between the pathological hyperphosphorylation of tau protein, a microtubuleassociated protein, and the formation of neurofibrillary tangles (NFT) [12]. The last decades had marked a very significant era of AD research. During this period, the nature of amyloid plaques and NTFs, the two histopathological hallmarks of AD, had been elucidated. Recent research efforts have led to several hypotheses to explain AD. Amyloid β toxicity is believed to play a primary role in the development of AD [13]. GSK-3β activity may increase with aging [14], which is consistent with the fact that aging is the most important risk factor for AD. Both in vitro and in vivo studies have demonstrated that inhibition of GSK- 3β, can reverse hyperphosphorylation of tau and prevent behavioral impairments in mice [15-20]. These studies make GSK-3β inhibition very attractive as a therapeutic target for AD [21].
We developed quantitative structure-activity relationships (QSAR) models for GSK-3β, linear discriminant analysis (LDA) [91] and linear artificial neural networks (ANNs) from more than 40000 cases with more than 24000 different molecules inhibitors of GSK-3β obtained from ChEMBL database http://www.ebi.ac.uk/chembldb/index.php/ target/browser/classification [92,93]; in total we used more than 45000 different molecules to develop the QSAR models. In addition, we did a study of different fragments that exist in the molecules of the database in order to see which fragments had more influence in the activity, and which fragments interact more with the protein. As there many studies with GSK-3β that can be found in the literature the design of new inhibitors of this enzyme is very important for the study of neurodegenerative diseases [94,95].
Linear classifier
A database from ChEMBL database [92] containing assayed GSK- 3β inhibitors was used (Table SM from the Supplementary Material). The DRAGON software 4.0 [14] was utilized here and provides 1664 descriptors classified as zero- (0D) one- (1D), two- (2D) and threedimensional (3D) descriptors depending on the fact they are computed from the chemical formula, substructure list representation, molecular graph or geometrical representation of the molecule, respectively [103]. In this work, we calculated the following descriptors: 2D autocorrelations, Burden eigenvalues, topological charge indices, eigenvalue-based indices, functional group counts, atoms-centred fragments, charge descriptors and molecular properties. The QSAR model was constructed with the multivariate regression technique, the LDA, employing the Forward stepwise method for the selection of variables. All statistical analyses and data exploration were carried out in STATISTICA 6.0 [104]. In the actual work, the independent data test is used by splitting the data randomly in a training series used for a model construction and a cross-validation (CV) one. The general formula of the QSAR classification function is the following:
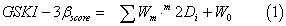
where GSKI-3βscore is the continuous and dimensionless score value for the GSKI-3β/non-GSKI-3β classification that gives relatively higher values to molecules with more probability to act as GSKI-3β, m2Di are the 2Ds of type m, Wm is the coefficient (weights) of these indices in the QSAR model and W0 is the independent term.
The reported statistical parameters of the QSAR model are the following: N, χ2, F, and p-level as well as Sensitivity, Specificity, and Accuracy for both training and CV [104]. N is the number of molecules used to train the model, λ is Wilks statistic parameter, χ2 is Chi-square and p-level is the probability of error.
Nonlinear classifiers
We processed our data with different ANNs using the STATISTICA 6.0 software [104] looking for a better model to predict activity against GSK-3β. Five types of ANNs were used, namely, Probabilistic Neural Network (PNN), Radial Basis Function (RBF) [105], Three Layers Perceptron (MLP-3), and Four Layer Perceptron (MLP-4) and Linear (LNN). The profile of a ANN is: Ni:I-H1-H2-O:No. It means that we have inputs variables (Ni), neurons in the input layer (I), neurons in the first hidden layer (H1), in the second hidden layer (H2), neuron in the output layer (O) and output variable (No).
We can used a very simple type of ANN called Linear Neural Network (RBF) to fit this discriminant function. The model deals with the classification of a compound set with or without affinity on different receptors. A dummy variable Affinity Class (AC) was used as input to codify the affinity. This variable indicates either high (AC = 1) or low (AC = 0) affinity of the drug by the receptor. S(DTP)pred or DTP affinity predicted score is the output of the model and it is a continuous dimensionless score that sorts compounds from low to high affinity to the target coinciding DTPs with higher values of S(DTP)pred and nDTPs with lowest values. In equation (6), b represents the coefficients of the RBF classification function, determined by the ANN module of the STATISTICA 6.0 software package [104]. We used Forward Stepwise algorithm for a variable selection.
Let be kχ(G) drugs molecular descriptors and kξ(R) receptor or drug target descriptors for different drugs (d) with different receptor; we can attempt to develop a simple linear classifier of mt-QSAR type with the general formula:
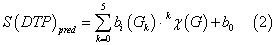
We assessed the quality of models with different statistical parameters like Specificity (Equation 2), Sensitivity (Equation 3), Accuracy (Equation 4) and ROC curve (Receiver Operating Characteristic curve) which is a graphical plot of the sensitivity, or true positives, vs. (1−specificity), or false positives,
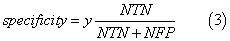
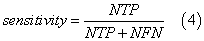
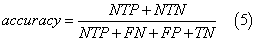
where NTN means number of true negatives, NFP is number of false positives, NTP is number of true positives, NFN is number of false negatives, FN is false negatives, FP is false positives and TN is true negatives.
The data set used in this article was obtained from ChEMBL database [92,93]. It has more than 56000 cases and more than 24000 different compounds inhibitors of GSK-3β. In total we used more than 45000 different molecules to develop the QSAR models obtained in ChEMBL. This is a database of bioactive drug-like small molecules, it contains 2-D structures, calculated properties (e.g. logP, Molecular Weight, Lipinski Parameters, etc.) and abstracted bioactivities (e.g. binding constants, pharmacology and ADMET data). ChEMBL normalises the bioactivities into a uniform set of end-points and units where possible, and also tags the links between a molecular target and a published assay with a set of varying confidence levels. The data is abstracted and curated from the primary scientific literature, and covers a significant fraction of the structure activity relationship (SAR) and discovery of modern drugs. The codes and activity for all compounds as well as the references used to collect them are depicted in Table SM of the supplementary material file.
LDA
In this paper we obtained a LDA study with Equation 6, and we can observe that eighteen variables entry inside equation:
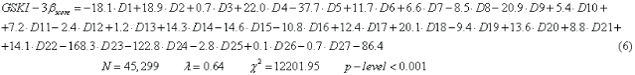
In Table 1, we show the code names of descriptors used in the equation 6. The nomenclature used in the descriptors of the equation is the same as establishing the Dragon software, where N is the number of compounds used for training, λ is the Wilks statistic parameter, χ2 is the Chi-square and p is the level of error. The model correctly classified 1310 out of 1643 active compounds (79.7%) and 24823 out of 26156 non-active compounds (94.9%) in the training series. The overall training performance was 94.0%. Validation of the model was carried out using an external predicting series. In this series the model classified correctly 757 out of 940 (80.5%) compounds and 14166 out of 14937 non-active compounds (94.8%). The overall predictability performance was 94.0% (Table 2).
| Definition | Name Descriptor |
|---|---|
| D1 | ATS1m |
| D2 | ATS2m |
| D3 | ATS8m |
| D4 | ATS3v |
| D5 | ATS3e |
| D6 | MATS3m |
| D7 | MATS4m |
| D8 | MATS3e |
| D9 | MATS2p |
| D10 | GATS1v |
| D11 | GATS4v |
| D12 | GATS1e |
| D13 | GATS7e |
| D14 | GATS3p |
| D15 | BELm4 |
| D16 | BELm5 |
| D17 | BELv2 |
| D18 | BEHe1 |
| D19 | BEHe8 |
| D20 | BELe5 |
| D21 | BELe8 |
| D22 | BELp4 |
| D23 | JGI4 |
| D24 | JGI7 |
| D25 | Ui |
| D26 | AMR |
| D27 | MLOGP |
Table 1: Code names of the different molecular descriptor used in the equation 6.
| Model | Train | Stat. | Validation | ||||
|---|---|---|---|---|---|---|---|
| profile | Active | Non-Active | % | Par. | % | Active | Non-Active |
| 1310 | 333 | 79.9 | Sn | 80.5 | 757 | 183 | |
| LDA | 1333 | 24823 | 94.9 | Sp | 94.8 | 771 | 14166 |
| 94.0 | Ac | 94.0 | |||||
| RBF | 1552 | 100 | 94.0 | Sn | 94.3 | 889 | 53 |
| 166:166-402-1:1 | 1572 | 25613 | 94.2 | Sp | 94.1 | 909 | 14611 |
| 94.0 | Ac | 94.2 | |||||
Table 2: Comparison of LDA and ANN classification model.
ANN models
The ANN models are non-linear models useful to predict the biological activity of a large datasets of molecules. This technique is an alternative to linear methods such as LDA [106,107]. Figure 6 depicts the networks maps for some of the ANN models. In general, at least one ANN of every types tested was statically significant. However, one must note that the profiles of each network indicate that these are highly nonlinear and complicated models [108-110].

Figure 6: Depicts the networks maps for some of the ANN models used in this manuscript.
In Figure 7, we depict the ROC-curve [111,112] for RBF tested. Notably, almost model presented and an area under curve higher than 0.5 (the value for a random classifier). The vitality of this type of procedures developing ANN-QSAR models has been demonstrated before [113]; see, for instance, the work of Fernandez and Caballero [114]. The same is true about the ANNs tested, where is illustrated ROC-curves of ANN RBF with an area higher than 0.99. To show how important is this result, we compared the present model with other model used to address the same problem. We processed our data with ANNs looking for a better model. In general, the ANN RBF tested was statically significant [107].
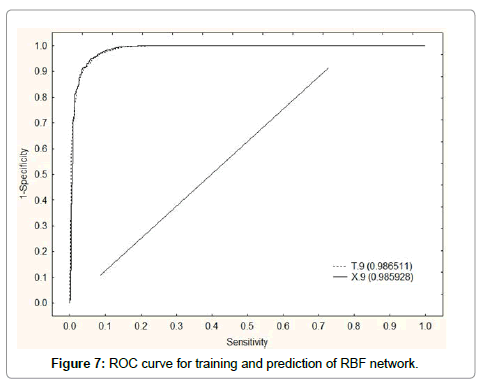
Figure 7: ROC curve for training and prediction of RBF network.
The network found was RBF and it showed training performance higher than 94.2%. The summary of results is showed in Table 2. After direct inspection of the results reported in Table 2 for ANN methods, we can conclude that a complex ANN method is a good method to predict the activity. We compare different types of networks to obtain a better model; Table 2 shows the classification matrix of the different networks. RBF 166:166-402-1:1 was taken as the main network because it presented a wider range of variables, 166 inputs in the first layer and 166 neurons in second layer, and two sets of cases (Training and Validation). Another tested networks found were LNN 233:233-1:1 and LNN 232:232-1:1 presented low accuracy and PNN 233:233-20619-2- 2:1 had a very low percentage of DTPs leading to possible errors in the model although its accuracy was very good, (Table 1). We depict the ROC-curve for RBF 166:166-402-1:1 to show how reliable was the network model developed, (Figure 7).
The functions of GSK-3 and its implication in various human diseases have triggered an active search for potent and selective GSK-3 inhibitors. Nowadays, theoretical studies such as QSAR models have become a very useful tool in this context to substantially reduce time and resources consuming experiments. In this work we developed a new LDA model using the Dragon descriptors, with a large data base using about 20000 different drugs obtained from the ChEMBL server. We conclude that a large database gives a much more precise model; the use of tools such as ChEMBL database enables us to develop models with large data bases, and this helps us to make the results more reliable. To improve the model we developed non-linear models and compared them to LDA. We proposed non-linear models, and for the first time, we proposed ANN models based on Dragon Descriptors series of GSK-3β, and we concluded that they are alternative methods to study the activity of different families of molecules compared with other methods found in the literature.
The authors thank the sponsorship DCS-UQROO PIFI (P/PIFI-2012- 23MSU0140Z-09 DCS) and FJPP thanks sponsorshipsfor a research position at the University of Quintana Roo from the project.