Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Review Article - (2016) Volume 4, Issue 3
Keywords: Reticulin fibrosis; Collagen fibrosis; Myelofibrosis; Myeloproliferative disorders; Myeloproliferative neoplasm; Polycythemia vera;
The term “myeloproliferative disorder” (MPD) is used to refer to those conditions in which there is a primary clonal stimulation of bone marrow hematopoietic stem cells producing erythroblasts, granulocytes and megakaryocytes [1]. The clonal evolution of MPD leads to hypercellularity of hematopoietic progenitor cells in the bone marrow either trilinear in classic PV, “unilinear” megakaryocytic in “true” ET or bilinear in MPD presenting with a dual chronic or primary megakaryocytic and granulocytic myeloproliferation (CMGM/PMGM) [1]. Such intrinsic stimulation or autonomous growth of clonal hematopoietic cells in the bone marrow as well as in the extramedullary sites-in particular the spleen-begins long before increase of reticulin and collagen fibers (myelofibrosis) does occur.
The resultant initial clinical picture begins with the appearance of enlarged megakaryocytes ranging from mature, pleiomorphic in ET and PV with increasing cellularity of erythropoiesis and/or granulopoiesis in the bone marrow and progressive hematopoietic neoplasia in the spleen. The trilinear MPDs labeled as myeloproliferative neoplasms (MPNs) in the 2008 WHO classification are associated with various degrees of thrombocytosis, erythrocytosis and granulopcytosis in the prefibrotic stages. Over a period of years splenic myeloid neoplasia preceeds and myelofibrosis supervenes as two independent but related and different serious complications in various MPN, CML and MPN/MDS [1-6].
The advanced or endstage illness in MPD/MPN is referred to as agnogenic myeloid metaplasia (AMM) or myeloid metaplasia with myelofibrosis (MMM) labeled as primary myelofibrosis in the PVSG and WHO classifications. Frequently, the preceding subclinical MPD phase is overlooked due to crude minimal PVSG/WHO criteria in textbooks and official recommendations for the diagnosis of ET, PV, AMM and Primary Idiopathic Myelofibrosis (PMF) (Tables 1 and 2).
| Classification of CMPDs comparing general terminology with own system and their occurrence among 3,933 BMB from CMPD patients | |||
| WHO | Textbooks | Hannover-Clasification | %1 |
| Primary disease | |||
| CML and subtypes | CML or CGL and subtypes | CML | 13.91 |
| CML.CT | |||
| CML.M | |||
| P.VERA | P.VERA | P.VERA | 17 |
| Thrombocythemia2 | Thrombocythemia2 | Thrombocythemia2 | 7.1 |
| CMGM | 16 | ||
| Pre-early fibrotic | |||
| Agnogenicmylenoid metaplasia(AMM) | Primary idiopathic myelofibrosis or agnogenic myeloid metaplasia | Advanceddisease | 33.3 |
| Increase of blasts increase of fibres | |||
| Unclassifiable | Unclassifiable | Unclassifiable | 12.6 |
| 1Percentage from 3,933, 2Primary or Idiopathic or essential Thrombocythemia. | |||
Table 1A: Hannover bone marrow classification of chronic myeloid leukemia (CML) and myeloroliferative disorders (MPD) [4].
| Typing of myelosclerosis and – fibrosis within the Hannover Classification of CMPD | |||
| Typing | Early myelo-sclerosis EMS | Myelosclerosis,myelofibrosis MS/MF | Advanced myelofibrosis AMF |
| Fiber quality | Reticulum | Reticulum plus Collagen | Collagen |
| Fiber pattern | Focal, patchy | Diffuse network with patches of Collagen | Scarring |
| Sinus walls | No | Minor sclerosis | Fibrosed and extended |
| Bone sclerosis | No | Rare | By definition |
| Haematopoiesis | Unchanged | Mostly unchanged | Mostly reduced |
Table1B: Grading of reticulin and collagen fibrosis in the 1990 Hannover Bone Marrow classification of the Ph-negative MPDs true ET, PV and CMGM and in Ph+-ET and CML [4].
| Grading reticulin fibrosis (RF)[11,22] |
Grading MF | Description of reticulin and collagen myelofibrosis (MF) in myeloproliferative disorders (MPD)[19-21] |
| Normal RF-0 |
N = 0 | No reticulinfibers, occasional individual fibers or focal areas with tiny amount of reticulinfiber network. |
| Slight increase RF 1 |
1 + | Fine reticulinfiber network throughout much of section and no course reticukinfibers. |
| Moderate increase RF 2 |
2 + + | Diffuse finereticuline networkwith focal collections of thick course reticulinfibers and no collagenisation. |
| Marked increase RCF 3 BM dry tap |
3 +++ | Diffuse and dense increase in reticulin with extensive intersections, and presence of collagen fibersand no osteosclerosis. |
| OS Dry tap | 4 sclerotic |
Diffuse and dense reticulin with with coarse bundles of collagen associated with significant osteosclerosis. |
Table 2: Grading of reticulin fibrosis (RF) [11,22]. Reticulin and collagen fibrosis (RCF) [1,17,18] and myelofibrosis (MF) [19-21] using Gomorri’s silver stain for reticulin and Mason’s trichrome stain for collagen.
Cytogenetic studies, isozyme markers and gene mutations studies (polymerase chain reaction: PCR) between 1969 and 1981 demonstrated that fibroblast proliferation in ET, PV, and AMM appeared to be polyclonal [1]. This simple indicates that the various degrees of MF (RF and RCF, Table 2) is a reactive process whereas the hematopoietic stem cells appeared to be of clonal origin in each of the chronic myeloproliferative disorders AMM (PMF), PV, ET and in CML [1]. The 1975 PVSG classification excluded Ph+ ET and CML from the Ph- negative MPDs ET, PV and MF [2,3]. The secondary stimulation of polyclonal fibroblasts by cytokines from clonal neoproliferative megakaryocytic and granulocytic precursors in the bone marrow gives rise to reticulin fibrosis (RF) or reticulin/collagen fibrosis (RCF) of the bone marrow as a serious irreversible complication, which depends on the aggressiveness and degree of the neoproliferative grade of “malignancy” of clonal hemotopoietic stem cells.
As MF advances to grade 3 or more, this ultimately will lead to bone marrow hypocellularity and anemia as a typical feature of the fibrotic stage of AMM or PMF usually associated with splenomegaly and hypersplenism.In the present analysis we evaluate the significance of secondary reticuline fibrosis (RF) and advanced reticulin-collagen fibrosis (RCF) in patients with PVSG-defined MPDs ET, PV and AMM (1986) in view of the Hannover Bone Marrow Classification[4-6] (1990), and the 2001 and 2008 WHO classifications for MPD and MPN respectively [7,8].
The supporting network of bone marrow is composed of branched fibroblastic cells that elaborate delicate fibers termed reticulin. Reticulin fibers (RF) are blackened by silver impregnation. In contrast, collagen fibers are courser, stain with Masson trichrome stains, and do not usually blacken with silver impregnation [9-11]. Biochemical methods and immunofluorescent staining indicate that fine reticulin fibers are mainly ‘type III collagen’ and the course collagen fibers are ‘type I collagen’9. The terms reticulin fibrosis and collagen fibrosis are well established in the literature [10,11]. In bone marrow fibrosis (myelofibrosis: MF), sequential biopsies indicate that initially there is a diffuse or patchy increase in fine reticulin fibers admixed with abundant hematopoietic elements (reticulin fibrosis in PV, ET and AMM or PMF, Figures 1 and 2). In later bone marrow biopsies, the marrow is replaced by course collagen fibers with decreased and a paucity of hematopoietic cells (collagen fibrosis, Figure 2). This progression from reticulin fibrosis to collagen fibrosis in the bone marrow biopsy during long-term follow-up may be analogous to the similar wound healing, in which collagen composition changes as time passes (fine reticulin type III collagen is replaced by course collagen type 1 collagen) [9]. The precise mechanisms by which cytokines from abnormal neoproliferative hematopoietic clone in the various MPDs do stimulate the host’s polyclonal fibroblasts to produce excessive amounts of fine reticulin fibers and course collagen bundles are under investigation and out of scope in this review.
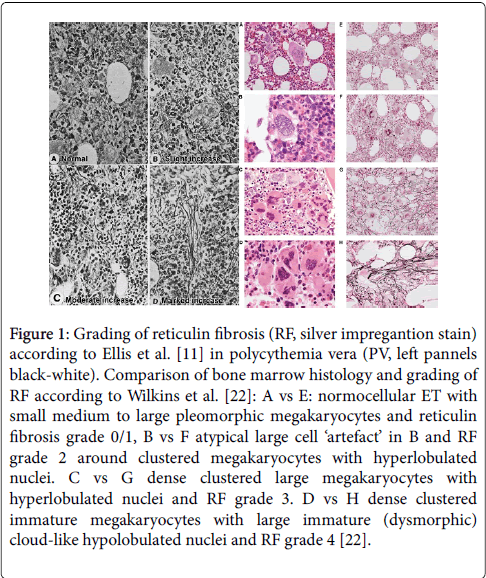
Figure 1: Grading of reticulin fibrosis (RF, silver impregantion stain) according to Ellis et al. [11] in polycythemia vera (PV, left pannels black-white). Comparison of bone marrow histology and grading of RF according to Wilkins et al. [22]: A vs E: normocellular ET with small medium to large pleomorphic megakaryocytes and reticulin fibrosis grade 0/1, B vs F atypical large cell ‘artefact’ in B and RF grade 2 around clustered megakaryocytes with hyperlobulated nuclei. C vs G dense clustered large megakaryocytes with hyperlobulated nuclei and RF grade 3. D vs H dense clustered immature megakaryocytes with large immature (dysmorphic) cloud-like hypolobulated nuclei and RF grade 4 [22].
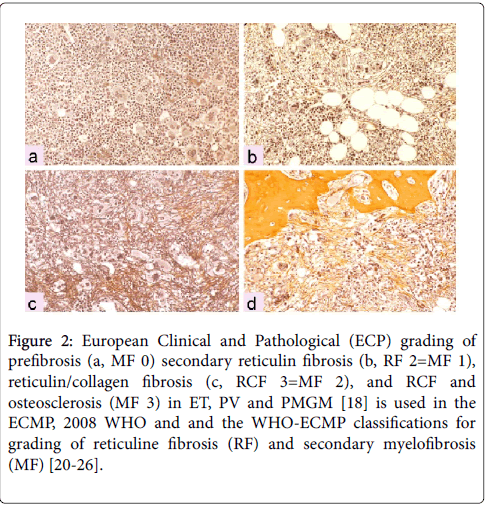
Figure 2: European Clinical and Pathological (ECP) grading of prefibrosis (a, MF 0) secondary reticulin fibrosis (b, RF 2=MF 1), reticulin/collagen fibrosis (c, RCF 3=MF 2), and RCF and osteosclerosis (MF 3) in ET, PV and PMGM [18] is used in the ECMP, 2008 WHO and and the WHO-ECMP classifications for grading of reticuline fibrosis (RF) and secondary myelofibrosis (MF) [20-26].
At least two kinds of fiber qualities can easily be distinguishes by common staining in light micsroscopy: reticulin fibrosis (RF) and collagen fibrosis (CF). Gommori’s silver staining detects early and course reticulin fibers (RF) and do not stain collagen fibers thereby underestimating advanced RCF myelofibrosis grade 2 and 3 (Table 2). Collagen fibers stain with a Mason’s trichrome stains. Silver stain does not distinguish RF from RCF in advanced myelofibrosis (MF) grade 2 and 3. Consequently both Gommorri’s stain for RF and trichrome stain for collagen fibrosis (CF) are to be used for optimal MF-grading of RF and RCF. The evolution of RF into RCF as documented by the combined use of silver and trichrome stains simple means a determinative change from reversible normal reticulin (+RF) into irreversible pathological collagen scarring (+RCF without or with osteosclerosis) (Figures 1 and 2).
Clinically, RCF often results in dry tap, when aspiration is attempted. Reticulin fibrosis grade 0/1 and RF with very early collagen fibrosis (RCF) usually do occur without real scarring. Bone marrow aspiration in RF without CF usually does not cause the symptom of dry tap. Advanced myelofibrosis (RCF = MF 2 and 3) designates a pronounced increased collagen fibrosis with visuable scarring spotted areas and sometimes with foci or larger areas of atrophic hematopiesis in the bone marrow in light microscopy. According to the Hannover Bone Marrow Classification the risk or probability of prefibrotic ET and PV to develop myelofibrosis grade 2/3 in sequential BM biopsies is very low in ET, rather low in PV but high in CMGM/PMGM (Figure 3) [1,6].
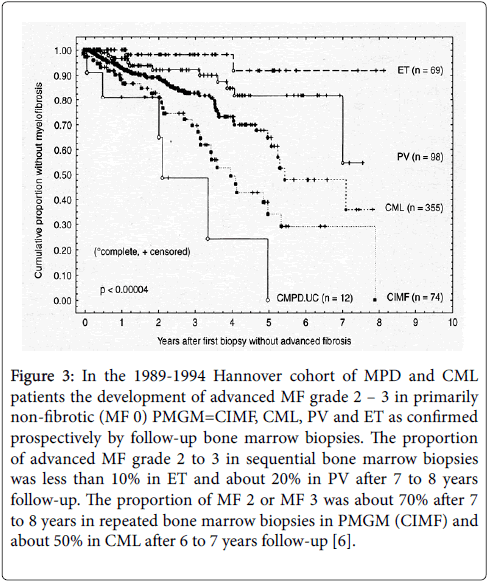
Figure 3: In the 1989-1994 Hannover cohort of MPD and CML patients the development of advanced MF grade 2 – 3 in primarily non-fibrotic (MF 0) PMGM=CIMF, CML, PV and ET as confirmed prospectively by follow-up bone marrow biopsies. The proportion of advanced MF grade 2 to 3 in sequential bone marrow biopsies was less than 10% in ET and about 20% in PV after 7 to 8 years follow-up. The proportion of MF 2 or MF 3 was about 70% after 7 to 8 years in repeated bone marrow biopsies in PMGM (CIMF) and about 50% in CML after 6 to 7 years follow-up [6].
A prospective bone marrow pathology study in newly diagnosed PV patients of the PVSG 01 study evaluated grading of cellularity and reticulin fibrosis in PV [11]. Characteristic findings in BM biopsies of 155 evaluated PV patients in the PVSG 01 study revealed a broad spectrum of no, slight, moderate to marked (˃80%) increase of bone marrow cellularity. Increased bone marrow cellularity of 60 to 100% was recorded in 145 of 155 PV patients [11].
The characteristic diagnostic feature for PV was the increase and clustering of large pleomorphic megakaryocytes in a hypercellular bone marrow (60-100%). Such pleomorphic megakaryocytes are also present but less pronounced when bone marrow cellularity was normal. In these BM biopsies of the PVSG 01study silver stained reticulin was graded according to Foot and Foot Masson’s trichrome stain was not used in this study (Table 2). In the PVSG bone marrow biopsy study, reticulin was normal (prefibrotic) in 61%, slightly increased (early fibrotic) in 26%, and moderately to marked increased in 13%.
At that time the finding of increased clustered enlarged megakaryocytes in a hypercellular bone marrow was known to be diagnostic for PV (reviewed by Michiels et al. 2006) [1]. From the prospective PVSG 01 bone marrow biopsy study in previously untreated PV patients it can be concluded that 79 of 94 (84%) of well documented PV patients have prefibrotic MPD with no or slight increase of RF. Only 13% of these PV patients have myelofibrotic disease with moderate or advanced RF.
Agnogenic myeloid metaplasia (AMM) or primary myelofibrosis (PMF)
In 1975 the PVSG defined the criteria for PV, primary hemorrhagic thrombocythemia (PTH) and primary myelofibrosis (PMF) or agnogenic myeloid metaplasia (AMM) as three distinct variants of MPD [2,3]. In 1977 Silverstein updated the spectrum of PVSG defined primary hemorrhagic thrombocythemia (PTH) versus AMM [12]. AMM is a clinicopathological entity not preceded by any other PVSG defined MPD including PTH, PV, CML, or preleukemia (MDS) [2,12] and characterized by various degrees of anemia, splenomegaly, leukoerythroblastosis, with tear drop-shaped erythrocytes, and dry tap on BM aspiration due to various degrees of myelofibrosis (MF) or osteomyelofibrosis. PMF or AMM patients are usually of age between 50 to 80 years, have enlarged spleens, a leukoerythroblastic blood reaction, striking teardrop poikilocytosis and dry tap on bone marrow aspiration.
According to Silverstein a typical PMF or AMM bone marrow is fibrotic in most cases, hypocellular in 85%, normocellular in 5% and hypercellular in 10%. In the studies of Thiele the mean age of advanced PMF or AMM.M is above 60 and around 70 years [13,14]. According to our experiences as documented in 1992 [15] masked MPD preceding PMF for 10 to 15 years must have been overlooked as the consequence of extremely crude criteria for AMM (PMF) of anemia, splenomegaly and myelofibrosis (Table 3). We could confirm the existence of primary myeloproliferative disease in several cases within our cohort of a few hundreds of MPD patients with various MPDs patients seen between 1975 and 2015. This is illustrated by the case history published in 1992 (Table 3) with primary myeloproliferative disease (PMD) not meeting the criteria of ET, PV, AMM or PMF, PMD was diagnosed by typical MPD features in the bone marrow, and a palpable spleen tip below the costal margin in a 61-year-old man with normal values for haemoglobin, hematocrit, platelet and leukocyte counts [15].
| Hematological Findings in cases of Erythromegalia and Atypical Thrombocythemia* | ||||||
| 1971 | 1978 | 1985 | 1988 | 1989 | 1989a | |
| Erthromegalia | - | - | + | + | + | - |
| Peripheral Blood | ||||||
| Hemoglobinmmol/l | 9.8 | 8 | 7.1 | 5.9 | 5.7 | 5.8 |
| Platelets × 109/l | 285 | 265 | 421 | 484 | 811 | 223 |
| Leukocytes × 109/l | 6.9 | 10.8 | 21.4 | 21.9 | 24 | 18 |
| Bone Marrow | ||||||
| Cellularity | N | ↑ | ↑ | ↑ | ||
| Erythropoesis | N | N | N | N | ||
| Ringed sideroblast | - | - | - | - | ||
| Megakaryocytes | ↑ | ↑ | ↑↑ | ↑↑ | ||
| Reticulin | ↑ | ↑↑ | ↑↑↑ | ↑↑↑ | ||
| Collagen | - | - | ↑ | ↑↑ | ||
| Osteoid | - | - | - | + | ||
| Myelodysplasia | - | - | - | - | ||
| Karyotype of bone marrow cells | ||||||
| Spleen size on scan | ||||||
| Length diameter cm | 18 | 23 | ||||
| *N:Normal; ↓: Decreased; ↑: Increased; - Absent; +:Present; one arrow slight two arrow pronounced and three exceptional. The peripheral blood and bone marrow findings are consistent with agnogenic myeloid metaplasia in case 1. aWhile on treatment with Hydroxyurea. | ||||||
Table 3: Long-term follow-up of a 61-year-old man, who presented in 1971 with primary myeloid metaplasia of the spleen initially diagnosed as prefibrotic primary myeloproliferative disiease (MPD). Due to hypersplenism related to increased spleen size. 18 cm to 23 cm length diameter on scan (normal value <12 cm) in 1978 and 1988, the peripheral blood counts remained normal for 14 years of follow-up. From 1985 to 1989 he suffered from aspirin responsive erythromelalgia of fingers and toes at platelet counts of 421×109/L. After 17 years of follow-up this case of PMD developed in 1988 anemia,thrombocythemia, and symptoms related to splenomegaly consistent with the diagnosis of agnogenic myeloid metaplasia and advanced myelofibrosis (MF 2/3) [27].
The 20 years follow-up was featured by a progressive myeloid metaplasia and myelofibrosis. Such cases of AMM has been described by Snapper in 1960 as aleukemic megakaryocytic myelosis [16]. This aleukemic megakaryocytic myelosis is characterized by proliferation of megakaryocytes in bone marrow spleen and liver is nearly always complicated by myelofibrosis and myelosclerosis. Clinical signs commonly consist of tremendous spleen, large liver, severe anemia and leukopenia and abnormally shaped red cell, so-called teardrop cells, myelocytes, myeloblasts and especially normoblasts are often seen in the peripheral blood [16]. In about one-third of cases the AMM develops in patients with a burnt-out polycythemia [16]. The diagnostic criteria PVSG defined PMF or AMM without features of PV at diagnosis and during follow-up have not been changed and persisted in 2001 WHO classification as CIMF [7] and in the 2008 WHO classification as PMF [8].
In 1990, Georgii et al. proposed the Hannover Bone Marrow (BM) Classification and separated at the bone marrow level the PVSG defined ET PV AMM into PV, ET, PV, and PMGM features at the bone marrow level [4]. The Hannover BM classification of the CMPDs, clearly distinguished ET (Thrombocythemia) and AMM from PMGM as the third disctinct MPD entity (Table 1) [4-6]. The Hannover BM classification defined three new groups of primary prefibrotic myeloproliferative disorders ET, PV and PMGM at the bone marrow level without use of clinical variables. The chronic MPDs according to the Hannover bone marrow classification do not use PVSG criteria. The Hannover bone marrow classification of the prefibrotic MPDs clearly distinguishes: 1) ET; 2) typical PV; 3) prefibrotic chronic or primary megakaryocytic granulocytic myeloproliferation (CMGM/ PMGM); 4) advanced MPD disease (similar to AMM) with increased RCF or blasts; and 5) MPD unclassifiable (Table 1, Figure 3) [4-6]. In our experiences a typical PV bone marrow picture is seen in PV, but also in ET mimicking PV (prodromal PV, which in later studies proved to be, EEC+ and may have low serum EPO [1,17,18]. In the Hannover Bone Marrow classification CMGM is featured by hypercellular ET associated with chronic or primary megakakaryocytic granulocytic myeloproliferation (CMGM [4-6] or PMGM) and clearly in between between normocellular ET and fibrotic AMM or PMF [7,8]. In the experience of Georgii the spectrum of mature large megakaryocytes in ET versus dysmorphic megakaryocytes in CMGM/PMGM is overlapping with a grey zone of about 10%. Pronounced dysmorphic megakaryocyte with immature with immature cloud/like nuclei in CMGM/PMGM are almost never seen in normocellular ET, prodromal PV or PV [17-27]. Post-PV myelofibrosis indeed may show pronounced pleomorphic megakaryocytes without typical cloud-like nuclei in fibrotic RCF stage of the PV or advanced ET disease. The clinically oriented PVSG categorization and classification of ET, PV and AMM do not show congruity with the Hannover bone marrow (BM) classification of the prefibrotic and fibrotic MPDs ET, PV and CMGM [18]. Consequently, the clinical PVSG and the Hannover bone marrow (BM) classifications should be used separately and independently from the PVSG clinical criteria for ET, PV and PMF.
A progress from MF 0 into RCF (MF 2) can be observed in the majority of chronic idiopathic myelofibrosis (CIMF = PMGM = PMF) patients indicating that PMGM or PMF is clearly in between true ET and AMM [18-23]. About one third of the PMGM patients, as evaluated by repeated BMB, remain free of RCF (= advanced MF 2, 3 = AMM). Most of the ET and PV patients did not proceed or only will proceed to RF (MF 1) without collagenization only (Figure 4, Table 8) [23].
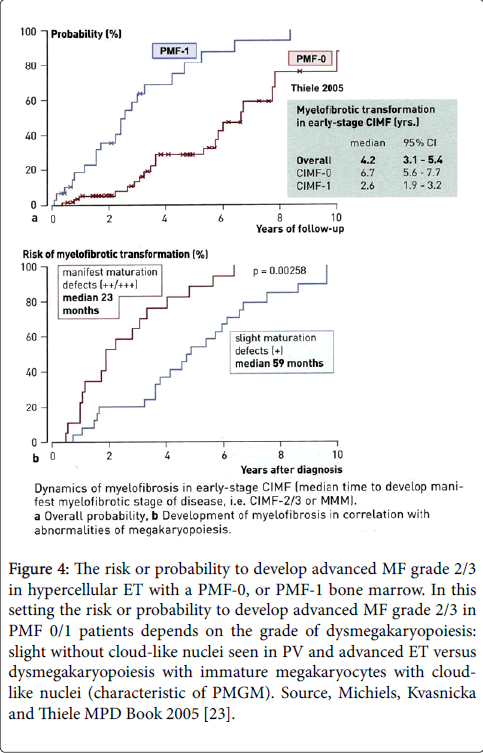
Figure 4: The risk or probability to develop advanced MF grade 2/3 in hypercellular ET with a PMF-0, or PMF-1 bone marrow. In this setting the risk or probability to develop advanced MF grade 2/3 in PMF 0/1 patients depends on the grade of dysmegakaryopoiesis: slight without cloud-like nuclei seen in PV and advanced ET versus dysmegakaryopoiesis with immature megakaryocytes with cloudlike nuclei (characteristic of PMGM). Source, Michiels, Kvasnicka and Thiele MPD Book 2005 [23].
There are cases of CIMF/PMF with slight dysmorphic megakaryocytes (no cloud-like nuclei) that cannot reliable be distinguished from ET (grey zone±10%) since platelet counts may be rather high in prefibrotic or early fibrotic (RF) (Figure 4) [23]. This means that PVSG defined prefibrotic or early fibrotic ET with a hypercellular MGM bone marrow indeed do show various degrees of dysmorphic megakaryocytes [17,18].
By further course of disease, the differences between CMGM (Table 1) or PMGM and ET will become obvious. CMGM/PMGM usually present with hypercellular ET with transient pronounced thrombocytosis (around or above 1000×109/L) followed by progressive decrease of platelet counts related to the degree of splenomegaly during follow-up [1]. In contrast ET is featured by persistent increase of platelet counts from low (400×109/L) followed by slow increase to values around and above 1000×109/L during long-term follow-up in normocellular ET and prodromal PV [1]. Fiber increase (RF or even RCF) is very rarely in ET, more frequent in PV and rather frequent in CMGM/PMGM [5,6]. All MPD.UC (not meeting the PVSG or WHO criteria) developed advanced MF 2, 3 (AMM or PMF (Figures 3 and 4).
There may be prefibrotic ET cases with increased cellularity due to increased erythropoiesis (prodromal PV). Such cases are diagnosed as PV in the Hannover Bone Marrow classification, which can be confirmed by the presence of EEC+ and low serum EPO according to the ECP criteria for PMD, prodromal PV and overt PV proposed by the Thrombocythemia Vera Study Group (TVSG) [17,18]. The UK MPD classification distinguishes 4 grades of reticulin fibrosis (RF) are defined (RF 1, 2, 3 and 4, Figure 1, Wilkins et al. [22]) while the 2001 WHO classification used the 4 grades of myelofibrosis (MF): MF-0, 1, 2 and 3 (Table 2) [1,7,20]. Thiele et al. confirmed that MF is not a feature of normocellular ET and very few WHO-ET patients will develop MF during long-term follow-up [19,23,24]. MF is present in only a minority of PV patients at diagnosis, but all stages of MF have been observed in PV during long-term follow-up in about one third of post-PV myeloid metaplasia [18-20].
According to the Cologne [19], ECP [20] and 2001 WHO [1,7] criteria the finding of all grades of MF (thrombocythemia associated with prefibrotic MF 0, early fibrotic MF 1 (false ET) versus overt fibrotic MF 2/3, AMM) is remarkable in the first biopsy of patients with CIMF (2001 WHO) or PMF (2008 WHO) (Figures 9 and 10) [14,24]. According to Thiele and Michiels PMF with MF 0 and MF 1 and AMM = PMF MF 2/3 (2008 WHO) is one continuous MPD disease [14,19-21,24-26]. With the advent of the JAK2V617F discovery by Vainchenker in 2005 [28], we immediately separated JAK2 mutated normocellular ET, prodromal PV ET with an MGM bone marrow histology from JAK2 wild type hypercellular CMGM/PMGM as distinct entities [1,25,26]. Please note, that masked ET, PV and PMF (asymptomatic or symptomatic) not meeting the PVSG or 2001 WHO diagnostic criteria) is rather frequent. Comparing the PVSG and WHO criteria Thiele and Kvasnicka demonstrated a low risk of MF evolution in normocellular ET (WHO-ET), but there is a higher risk of myelofibrotic transformation in JAK2 wild type CMGM/PMGM or PMF from the prefibrotic (PMF 0) or early fibrotic stage (PMF 1) to overt (PMF 2/3 or AMM).
This last stage meets the WHO criteria for PMF [14,23-25]. This progression of PMF or CMGM/PMGM disease as determined by bone marrow fibrosis is accompanied by a change into dysmorphic megakaryopoiesis with manifest maturation defects of cytoplasm and nuclei both in JAKV617F positive and JAK2 wild type AMM [1]. It is very well known that JAK2V617F mutated prefibrotic hypercellular ET with a PV bone marrow picture due to increased erythropoiesis (prodromal PV) or increased megakaryocytic granulocytic myeloproliferation (masked PV) and in JAK2 wild type CMGM/ is rather frequent and clearly differ from normocellular ET at the clinical and bone marrow levels [14,23-25].
The evolution of myelofibrosis grade 2 and 3 in JAK2 mutated masked PV and in JAK2 wild type CMGM/PMGM is accompanied by dysmegakaryopoiesis with the development of more pronounced dysmegakaryopoiesis, slowly progressive anemia, splenomegaly, increase of circulating CD34 cells and leukoerythroblastosis. Please note that patients diagnosed as fibrotic PMF = AMM = MMM with no adverse signs still may have survival times of another 10 years. Consequently, risk stratification according to Dupriez, Cervantes or IPSS is warranted for all stages of MF in PV and CMGM/PMGM using the CART model on top of the COX model of prognosis assessment within each stage of CIMF/PMF as compared to true ET (Figure 5) [14].
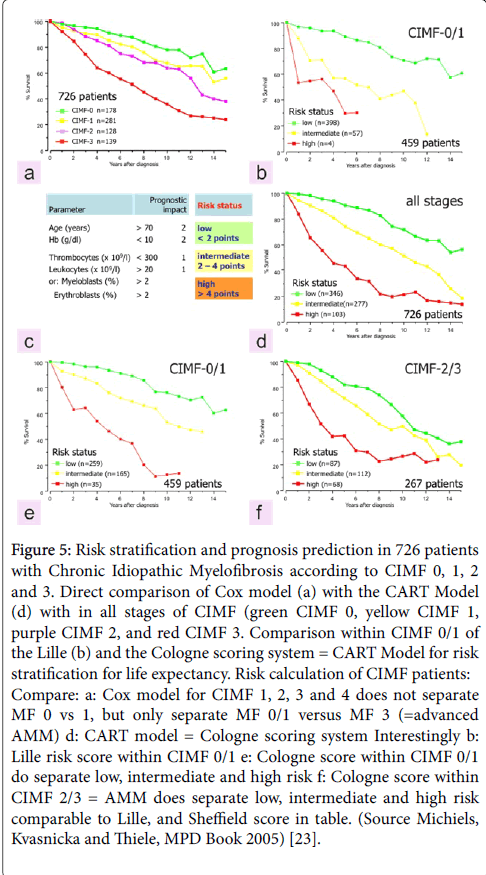
Figure 5: Risk stratification and prognosis prediction in 726 patients with Chronic Idiopathic Myelofibrosis according to CIMF 0, 1, 2 and 3. Direct comparison of Cox model (a) with the CART Model (d) with in all stages of CIMF (green CIMF 0, yellow CIMF 1, purple CIMF 2, and red CIMF 3. Comparison within CIMF 0/1 of the Lille (b) and the Cologne scoring system = CART Model for risk stratification for life expectancy. Risk calculation of CIMF patients: Compare: a: Cox model for CIMF 1, 2, 3 and 4 does not separate MF 0 vs 1, but only separate MF 0/1 versus MF 3 (=advanced AMM) d: CART model = Cologne scoring system Interestingly b: Lille risk score within CIMF 0/1 e: Cologne score within CIMF 0/1 do separate low, intermediate and high risk f: Cologne score within CIMF 2/3 = AMM does separate low, intermediate and high risk comparable to Lille, and Sheffield score in table. (Source Michiels, Kvasnicka and Thiele, MPD Book 2005) [23].
The clinical and bone marrow histology features of “true” ET has been described by Thiele et al. in 1988 and 2005 as distinct from PV (Figure 6) [28,29]. The peripheral blood findings in ‘true’ ET are featured by high platelet counts, normal values for haemoglobin, haematocrit, erythrocyte, white blood cells, LAP score, LDH and no or minor splenomegaly despite platelet counts above 1000×109/L (Figures 6 and 7)[28,29]. In 2002 Michiels and Thiele defined ‘true’ ET (Table 4) and differentiated “true” ET from hypercellular ET associated with prefibrotic CIMF or primary megakaryocytic granulocytic myeloproliferation (PMGM, Table 5)[20]. According to the 2002 ECP criteria in Table 4 “true” ET display large to giant megakaryocytes showing hyperlobulated staghorn-like nuclei in a normocellular bone marrow (Figures 6 and 7) [20].
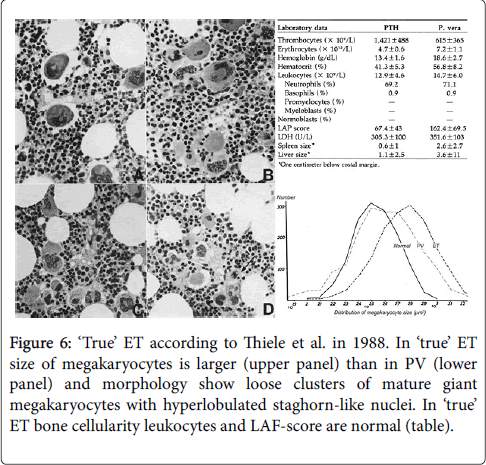
Figure 6: ‘True’ ET according to Thiele et al. in 1988. In ‘true’ ET size of megakaryocytes is larger (upper panel) than in PV (lower panel) and morphology show loose clusters of mature giant megakaryocytes with hyperlobulated staghorn-like nuclei. In ‘true’ ET bone cellularity leukocytes and LAF-score are normal (table).
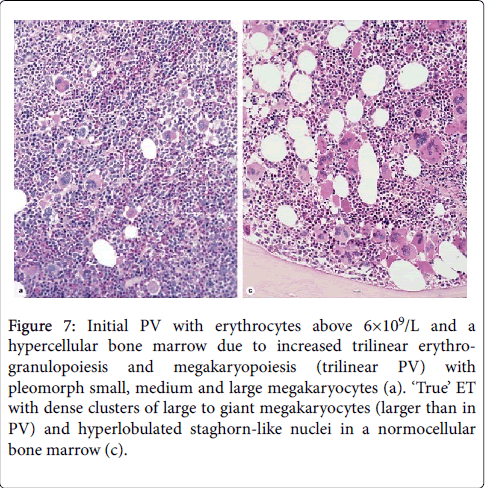
Figure 7: Initial PV with erythrocytes above 6×109/L and a hypercellular bone marrow due to increased trilinear erythrogranulopoiesis and megakaryopoiesis (trilinear PV) with pleomorph small, medium and large megakaryocytes (a). ‘True’ ET with dense clusters of large to giant megakaryocytes (larger than in PV) and hyperlobulated staghorn-like nuclei in a normocellular bone marrow (c).
| Clinical criteria | Pathological criteria | ||
| A1 | Persistent increase of platelet count: grade I: 400-1500, II: ˃1500×109/L | B1 | Predominant proliferation of enlarged megakaryocytes with hyperlobulated nuclei and mature cytoplasm, lacking conspicuous cytological abnormalities |
| A2 | Normal spleen or only minor splenomegaly on echogram | B2 | No proliferation of immaturity of granulopoiesis or eryhropoiesis |
| A3 | Normal Lap score, normal ESR, and increased MPV | B3 | No or only borderline increase in reticulin |
| A4 | Spontaneous megakaryocyte colony formation(CFU-Meg) | ||
| A5 | No signs or cause of RT | ||
| A6 | No preceding or allied other subtype of MPD, CML, or MDS | ||
| A7 | Absence ofthe Philadelphia chromosome | ||
| *the combinations of A1 and B1+B2 establish (true) ET (thrombocythemiavera).Any other criterion confirms ET. LAP indicates leukocyte alkaline phosphatse; ESR, erythrocyte sedimentation rate; MPV, mean platelet volume; CRU-Meg, colony-forming unit –megakaryocyte; RT, reactive thrombocytosis; MPD, myeloproliferative disorder; CML, chronic mylenoid leukemia; MDS, myelodysplastic syndrome. | |||
Table 4: The European Clinical and Pathological (ECP) criteria for ‘True’ ET [20].
PV was typically featured by large pleomorphic megekaryocytes with hyperploid nuclei in a hypercellular bone marrow due to increased erythropoiesis or increased erythrocytic-megakaryocyticgranulocytic megakaryocyticgranulocytic myeloproliferation. Interestingly the megakaryocytes in “true” ET were larger than in classical PV (Figures 6 and 7).
According to 2002 ECP criteria in Table 5, hypercellular ET associated with prefibrotic CIMF (“false” ET) or PMGM is dominated by an increase of clustered atypical dysmorphic megakaryocytes due to increases of cellular and nuclear size and bulky nuclei with clumsy lobuli and irregular roundish shaped form (so-called cloud-like nuclei, Figure 8) [4-6,28,29], which are never described in JAK2V617F mutated ET and PV (Figures 9 and 10) [25,26].
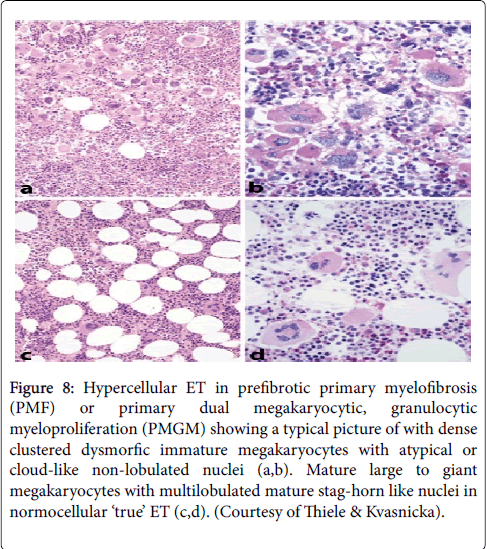
Figure 8: Hypercellular ET in prefibrotic primary myelofibrosis (PMF) or primary dual megakaryocytic, granulocytic myeloproliferation (PMGM) showing a typical picture of with dense clustered dysmorfic immature megakaryocytes with atypical or cloud-like non-lobulated nuclei (a,b). Mature large to giant megakaryocytes with multilobulated mature stag-horn like nuclei in normocellular ‘true’ ET (c,d). (Courtesy of Thiele & Kvasnicka).
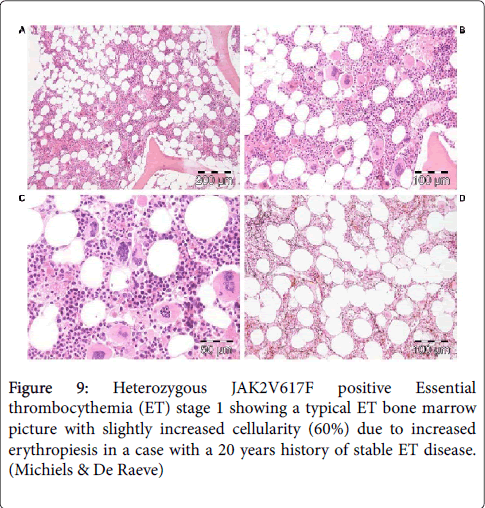
Figure 9: Heterozygous JAK2V617F positive Essential thrombocythemia (ET) stage 1 showing a typical ET bone marrow picture with slightly increased cellularity (60%) due to increased erythropiesis in a case with a 20 years history of stable ET disease. (Michiels & De Raeve)
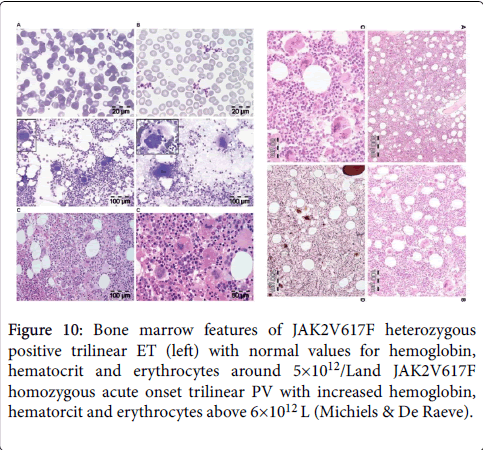
Figure 10: Bone marrow features of JAK2V617F heterozygous positive trilinear ET (left) with normal values for hemoglobin, hematocrit and erythrocytes around 5×1012/Land JAK2V617F homozygous acute onset trilinear PV with increased hemoglobin, hematorcit and erythrocytes above 6×1012 L (Michiels & De Raeve).
Bone marrow histopathology in patients with WHO-CMP defined prefibrotic JAK2V617F mutated normocellular ET or prodromal PV mimicking ET (Figures 9) and PV (Figure 10) are not different [25,26]. The pleomorphic megakaryocytes in JAK2V617F mutated ET in bone marrow smears and bone marrow biopsy were not larger but similar in size and pleomorphy with the large megakaryocytes in PV. The presence of clustered small and giant megakaryocytes with deeply lobulated stag-horn like nuclei (Figure 10) in normocellular ET carrying the MPL515 mutation are not seen in JAK2V617F positive ET, prodromal PV and PV (Figures 9 and 10).
There was local increase of erythropoiesis in areas of loose clustered pleiomorphic megakaryoctyes in JAK2V617F mutated normocelluar ET and in prodromal PV (Figure 9). JAK2 wild type ET carrying the MPL515 mutation have no clinical, laboratory and bone marrow features of prodromal PV at diagnosis do not evolve into PV during follow-up, and have normal LAP score, serum EPO and ferritin levels and therefore can readily be labeled as ‘true’ ET (Table 4).
Laboratory evaluation and bone marrow histology have the diagnostic potential to separate the JAK2V617F mutated ET and prodromal PV with increased LAP score, low serum EPO and pleomorphic megakaryocyte morphology from JAK2 wild type MPL515 mutated ET with normal LAP score and serum EPO and giant megakaryocytes with staghorn-like nuclei as clearly defined by Michiels and Thiele in 2002 (Figure 6, Table 4) [20].
The molecular etiology of JAK2/MPL wild type ET and MF remained elusive untill two Kralovics from Austria and Green UK independently discovered the calreticulin (CALR) mutations in MPN patients with JAK2 wild type ET or MF [30-32]. Dr Kralovics and his team in Austria and Italy described the occurrence of Calreticulin (CALR) mutation in 78 of 311 (25%) ET patients and in 72 of 203 (35%) MF patients and in none of 382 PV patients [30]. CALR mutations are mutually exclusive with both
JAK2V617F and MPL515 mutations. In none of 45 CML, 73 MDS 64 CMML and 24 RARS-T patients the CALR mutation was not found except that 3 SF3B1 positive RARS-T patients with myelodysplasia carried a CALR mutation [30,32]. Green and his team found somatic CALR mutations in 70 to 84% of MPN samples with nonmutated JAK2 ET or MF [31]. CALR exon 9 mutations were detected in 26 of 31 (84%) patients with JAK2 wild type ET or MF. CALR exon 9 mutations were absent in all 120 patients who had JAK2 or MPL mutations.
CALR mutations were present in 110 of 158 MPN patients lacking JAK2 or MPL, including 80 of 112 (70%) ET patients, 18 of 32 (56%) MF patients and 12 of 13 patients with progression of ET to MF. CALR mutations were identified in 10 of 120 (8%) MDS patients (RA in 5 of 53, RARS in 3 of 27 and RAEB-T in 2 of 27), and in one patient each with chronic myelomonocytic leukemia (CMML) and atypical CM [31]. Absence of PV laboratory features and no polycythemic transformation has been observed in CALR mutated patients [32]. Bone marrow histology in CALR mutated JAK2 wild type ET and MF in six consecutive CALR positive ET cases the bone marrow histology findings were consistent with hypercellular ET as the presenting feature of prefibrotic and early fibrotic stages of PMGM [32,33]. Bone marrow histology in prefibrotic CALR ET and early fibrotic CALR MF (JAK2 wild type) show dysmorphic megakaryocytes with definite abnormalities of maturation with bulky (bulbous) hyperchromatic nuclei and some disturbances of the nuclear cytoplasmic ratio (Figures 11 and 12) consistent with JAK2/MPL wild type PMGM (Table 5), which are not seen in MPL515 mutated ET and also not in JAK2V617F mutated ET, prodromal PV and PV. The bone marrow findings in JAK2/MPL wild type CALR mutated ET and MF (Figures 11 and 12) do not show features of PV in blood and bone marrow and are different from giant megakaryocytes and hyperlobulated staghorn-like nuclei in MPL515 positive ET (Figure 11).
| Clinical criteria | Pathological criteria | ||
| A1 | No preceding or allied other subtype of MPD,CML, od MDS | B1 | Megakaryocytic and granulocytic myeloproliferation and relative reduction of erythroid precursors. Abnormal clustering and increase in atypical gaint to medium-sized megakaryocytes containing bulbous (cloud-like) hypolobulated nuclei and definitive maturation defects. |
| A2 | Early clinical stages | ||
| Normal haemoglobin or anaemia, grade I: haemoglobin ≥12g/dL. | |||
| Slight or moderate splenomegaly onpalpationor >11 cm on ultrasoundscan or CT Thromobocythemi,platelets >400 ×109. | |||
| A3 | Intermediate clinical stage | ||
| Anemia grade II: haemoglobin ≥10g/dL | |||
| Definitive leuko-erythroblast blood picture and/or tear-drop erythrocytes | |||
| Splenomegaly | |||
| No adverse signs† | |||
| A4 | Advanced clinical stage | ||
| Anemia grade III: haemoglobin < 10g/L | |||
| One or more adverse signs† | |||
Table 5: The European Clinical and Pathological (ECP) criteria for prefibrotic chronic idiopathic myelofibrosis (CIMF) or primary megakaryocytic granulocytic myeloproliferation [20].
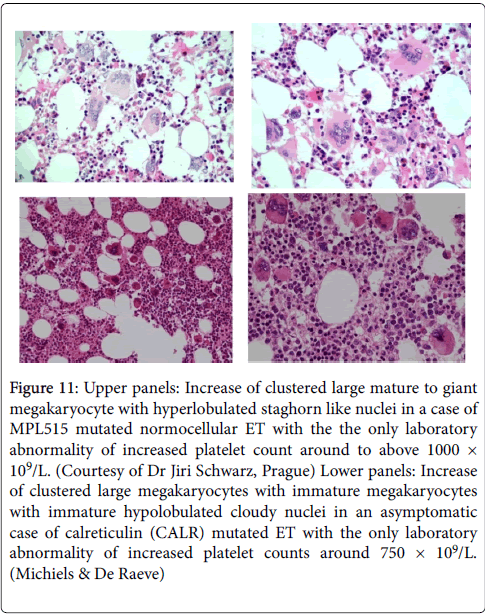
Figure 11: Upper panels: Increase of clustered large mature to giant megakaryocyte with hyperlobulated staghorn like nuclei in a case of MPL515 mutated normocellular ET with the the only laboratory abnormality of increased platelet count around to above 1000 × 109/L. (Courtesy of Dr Jiri Schwarz, Prague) Lower panels: Increase of clustered large megakaryocytes with immature megakaryocytes with immature hypolobulated cloudy nuclei in an asymptomatic case of calreticulin (CALR) mutated ET with the only laboratory abnormality of increased platelet counts around 750 × 109/L. (Michiels & De Raeve)
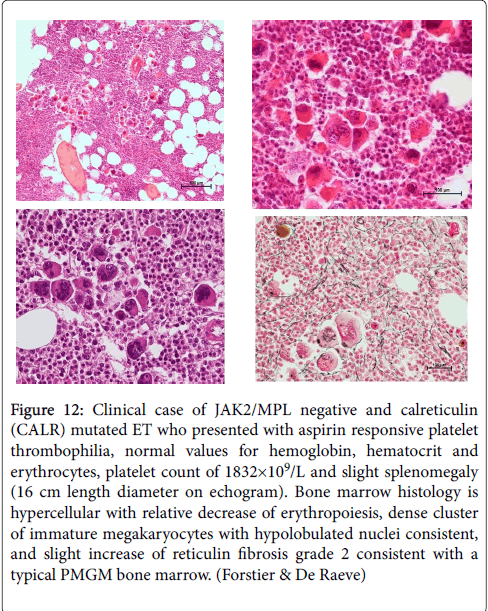
Figure 12: Clinical case of JAK2/MPL negative and calreticulin (CALR) mutated ET who presented with aspirin responsive platelet thrombophilia, normal values for hemoglobin, hematocrit and erythrocytes, platelet count of 1832×109/L and slight splenomegaly (16 cm length diameter on echogram). Bone marrow histology is hypercellular with relative decrease of erythropoiesis, dense cluster of immature megakaryocytes with hypolobulated nuclei consistent, and slight increase of reticulin fibrosis grade 2 consistent with a typical PMGM bone marrow. (Forstier & De Raeve)
The phenotypes of ET in various MPDs (MPNs) according to bone marrow features based on cellularity and megakaryocyte morphology and molecular etiology can be diagnosed as JAK2V617F-postive+ normocellular ET and ET with features of PV in the bone marrow (prodromal PV, JAK2 wild type normocellular ET carrying the MPL515 mutation [26,30-32]. JAK2, MPL and CALR mutated MPNs mutually eclude each other. The CALR mutated hypercellular ET and MF with a PMGM bone marrow histology will become the third distinct MPN entity [14,23-27]. A main unsolved conflict of the 2008 WHO classification regards the fact that the early fibrosis in the natural history of PMF is not defined in the 2008 WHO classification.
In the studies of Thiele normocellular ET and hypercellular ET with a prefibrotic MF bone marrow picture (pMF) irrespective of their molecular etiology did not differ significantly with regard to their clinical manifestation of MPN-specific microvascular thrombosis and bleeding and are to be treated similarly. Of course their natural histories surely will differ in terms of progression to myelofibrosis during long-term follow-up of 10 to more than 20 years. As show by Thiele and in many other studies, there is more than 65% probability of progression from prefibrotic (pPMF) into full-blown PMF. The degree of RF and MF proved to be unsuitable for use to stage MPN according to Lille, Cologne, IPSS, DIPSS and DIPSS-plus risk score assessment to predict prognosis and outcome [1,26]. The progression of fibrotic MPN disease is accompanied by a change of prominent clinical features. First, the development of anemia, splenomegaly and a leukoerythroblastic blood picture in the course of disease evolution is associated with a progressive change of pleiomorphic into dysmorphic megakaryopoiesis [4-6]. A confounding unsolved conflict is the definition of normocellular (true) ET by Thiele in all his publications before and beyond the 2008 WHO classification. True ET is featured by persistently increased platelet count, completely normal values for hemoglobin and hematocrit (around 0.41), slight leukocytosis but normal LAF score, and a normal cellular bone marrow without any increase of erythropoisesis or granulopoiesis [28,29]. This definition does not fit with the blood and bone marrow features of JAK2V617F positive ET, and excludes prodromal PV [32,33]. Interestingly, the platelets in “true” ET are large than normal (giant) [26] as compared to large pleomorphic megakaryocytes in JAK2V617F positive ET and PV. In our experience patients with prefibrotic JAK2V617F positive ET prodromal PV do have an increased LAF score, and a decreased serum erythropoietin levels (EPO) [1]. According to our recent experiences the 2002 ECP defined ‘true’ ET in Table 4, the laboratory and bone marrow features of normocellular ET with normal LAP score and normal serum EPO levels fits nicely with the findings in JAK2 wild type true ET carrying the MPL515 mutation. JAK2V617F positive ET and prodromal PV are typically featured by increased LAP score, decreased serum EPO with the presence of spontaneous endogenous colony formation (EEC).
With the advent of the JAK2V617F, MPL515 and CALR mutation, a third problem in the current WHO classification became evident by the demonstration of two variants of PMF MF 0 and MF 1 either JAK2V617F positive or JAK2 wild type CALR mutated ET and MF. This made the WHO classifications very confounding. The world-wide used criteria for fibrotic MPD disease as AMM by the 1975 PVSG [3], as CIMF by the 2001 WHO [7] and as PMF by 2008 WHO [8] classification detect the same cohort of primary advanced myeloid metaplasia with myelofibrosis grade 2 and 3 (AMM or PMF) and do mix up PMF with post ET and post-PV myelofibrosis of various clonal origin. All MPD classifications (1975-1986 PVSG, 2001 WHO and 2008 WHO) detect the same cohort of classical PV patients simple because they all used the same crude criterion of increased red cell mass, or equivalent high cut off levels for hemoglobin, hematocrit and/or erythrocytes (˃6×109/L) irrespective of serum ferritin level, serum EPO levels or EEC positivity. However 95% of WHO PV carry the JAK2V617F mutation [1]. A very important unsolved conflict of the 2001 and 2008 WHO classification is that all AMM or PMF patients must be preceded by a silent or symptomatic prodromal stages of primary myeloproliferative disease (PMD, Table 3). These early stages do not meet the PVSG, 2001 WHO and the 2008 WHO criteria. Early stage ET are overlooked by using a minimum platelet count of 600×109/L (PVSG, 2001 WHO) and 450×109/L (2008 WHO). This comprises about one third of PMD patients by the use of the very crude WHO inclusion/exclusion criteria for ET, PV and MF thereby overlooking masked ET, PV and PMD.
The 2015 WHO-CMP criteria define five clonal mutually exclusive myeloproliferative neoplasms: JAK2V617F mutated ET, prodromal PV, classical PV; JAK2 exon 12 mutated idiopathic erythrocythemia (IE) and PV; MPL515 mutated JAK2 type ET and MF; CALR mutated ET and MF and triple negative ET and MF of elusive etiology. Each of the five clonal MPN show distinct bone marrow features at diagnosis and during follow-up [1,26,32,33]. Pretreatment bone marrow histopathology findings in newly diagnosed MPN patients are of diagnostic significance for the differentiation between JAK2 mutated trilinear MPN versus MPL and CALR mutated ET and MF. Bone marrow histology in JAK2V617F mutated MPN patients (ET and PV) are similar and featured by clustered medium to large pleomorphic megakaryocytes and increased cellularity (60% to 90%) due to increased erythropoiesis. MPL515 mutated ET or ‘true’ ET is a distinct clonal MPN entity with large to giant mature megakaryocytes and hyperlobulated staghorn-like nuclei in a normocellular bone marrow without features of PV. Bone marrow histology in Calreticulin (CALR) mutated hypercellular ET revealed characteristic features of primary megakaryocytic, granulocytic myeloproliferation (PMGM) with dysmorphic immature megakaryocytes and cloud-like nuclei, which are not seen in MPL515 mutated ET and JAK2V627F mutated ET and PV [26,33,34]. On top of MPN symptoms and thrombosis burden, MPN disease burden is best reflected by the degree of anaemia, splenomegaly, bone marrow cellularity and increase of reticulin fibrosis [33,34]. Prospective studies are warranted to better define each of the pathophysiology of thrombocythemia phenotypes in the various distinct clonal MPNs at the clinical, laboratory molecular and bone marrow level of ET, PV and MF patients.