Advanced Techniques in Biology & Medicine
Open Access
ISSN: 2379-1764
ISSN: 2379-1764
Research Article - (2015) Volume 3, Issue 1
Background: Gal-32 is a Chinese hamster lung cell nuclear mutant that is unable to grow in galactose due to a defect in mitochondrial protein synthesis. Since the product of the Gal-32 gene was unknown, it was imperative to use phenotypic complementation to clone a human gene that corrected the Gal-32 mutation.
Results: Recessive Gal-32 cells were co-transformed with pSV2-neo plasmid DNA and recombinant DNA from a human genomic library containing the dominant human Gal+ gene and a chloramphenicol-resistance (camr) gene present in the pSV13 vector. Primary transformants were selected by growth in galactose and the neomycin analog G418. In order to rescue the human Gal+ gene, a genomic library was constructed with primary transformant DNA and the pCV108 cosmid vector. The camr gene was used to identify clones with the nearby human sequences. DNA from two camr, Alu-hybridizing clones was able to transform the recessive Gal-32 cells to the Gal+ phenotype and to restore mitochondrial protein synthesis.
Conclusion: These data demonstrate the isolation of two pCV108-transformant recombinant clones containing a human gene that complements the Chinese hamster Gal-32 mutation and restores galactose metabolism.
Keywords: Chinese hamster, Gal 32, Cosmid vector, HeLa cell
camr – Chloramphenicol resistance; CHL – Chinese Hamster Lung; CO – Cytochrome Oxidase; CR – Recombinant Clone; CsCl – Cesium Chloride; GAL – Galactose; Gal+ gene – confers ability to metabolize galactose; Gal- 32 – Galactose-deficient (inability to metabolize galactose); LB-Luria Broth; ND – NADH dehydrogenase; MXHAT - complete medium containing 78 mMmycophenolic acid (M), 1.6 mMxanthine (X), 0.1 mMhypoxanthine, 4.5 μMaminopterin(A), 40μMthymidine; Mt – Mitochondrial; pCV108 – cosmid vector containing the ampr and SV-neo genes; pSV2-neo – plasmid containing the gene that confers resistance to the neomycin analog G418; pSV13 – cosmid vector containing genes for chloramphenicol resistance (camr) and xanthineguanine phosphoribosyl-transferase (SV2-gpt); TR – Transformant
Gal-32 in an unusual Chinese hamster lung (CHL) cell mutant that lacks the ability to utilize exogenous galactose or fructose in place of glucose due to a single, recessive mutation that causes deficiencies in the mitochondrial (mt) respiratory chain [1,2]. Cells in culture growing in galactose or fructose obtain nearly all of their energy from glutamine via the Krebs cycle, the mt electron transport chain, and oxidative phosphorylation [3]. Therefore, cells that are defective in oxidative energy production by the mt electron transport chain fail to grow in galactose or fructose.
In order to grow or proliferate, cells must comply with the energy demand imposed by vital processes that include macromolecule biosynthesis, DNA replication, ion gradients generation and cell structure maintenance. Mitochondria, in general, play an important role in energy metabolism as they synthesize most of the cellular ATP through oxidative phosphorylation [4].
The mitochondrial genome encodes only 13 polypeptides, all of which are mitochondrially synthesized respiratory complex units [5,6]. However, most mitochondrial proteins, including most respiratory complex subunits, are nuclearly encoded and cytoplasmically synthesized. In Gal-32, the levels of mitochondrially synthesized proteins are all decreased to different extents unrelated to protein size; furthermore, there is a direct correlation between the activities of the respiratory chain and their corresponding mt translation products [7]. For example, NADH dehydrogenase (ND) activity and levels of mt encoded subunits ND 3, 4, 4L, and 5 are greatly reduced. Similarly, cytochrome c oxidase (CO) activity and mt synthesized subunits CO I, II, and III are drastically decreased. On the other hand, ATPase activity and mt encoded subunits 6 and 8 are only slightly altered. Likewise, succinate-cytochrome c activity, which is dependent on cytochrome b, and mt encoded apocytochrome b are marginally reduced. This differential reduction in mitochondrially synthesized proteins is a unique property of Gal-32 compared to other mammalian cell and yeast mutants, which usually exhibit no mt translation products [8-10].
Experiments investigating protein degradation as well as steady state levels and sizes of mt transcripts have revealed that the primary defect in Gal-32 is not caused by increased differential degradation of mitochondrial translation products, increased differential degradation of mtRNAs, or less efficient processing of the polycystronic precursor mtRNAs. Surprisingly, the steady state levels of both heavy and light strand mtDNA transcripts were elevated in Gal-32. Therefore, the differential reduction in mitochondrially encoded protein in Gal-32 seems to be the result of a decreased translation of specific mRNAs [11].
The differential decrease in mitochondrially synthesized proteins in Gal-32 is due to a single mutation as evidenced by the restoration to near normal levels of all mitochondrially encoded proteins and the respiratory complex activities in a spontaneous revertant that has regained the ability to grow on galactose. Using somatic cell hybrids, we have conclusively demonstrated the nuclear origin of the Gal-32 mutation. Fusion of rhodamine-6G-treated Gal+ cells with Gal-32 cells resulted in tetraploid hybrids that grew in galactose; this is expected for a nuclearly encoded gene since rhodamine-6G specifically inactivated mtDNA.
Since the product of the Gal-32 gene is unknown, it is necessary to use phenotypic complementation to clone the gene that corrects the Gal-32 mutation. The dominant wild type Gal+ 32 gene was transferred into recessive Gal-32 cells and phenotypic expression of the wild-type gene was selected for by growth of the transformants in galactose. In this paper, we report the successful isolation of two genomic cosmid clones containing a human gene that complements the Chinese hamster Gal-32 mutation. A preliminary report of this data was presented at a FASEB meeting [12].
The Howard University IRB (Internal Review Board) approved the use of the cell lines described herein and determined the protocol to be exempt based on 45 CFR 46.101(b)(4) and involves minimal risk. The human gene used in this study was derived from the HeLa cell line, which is available commercially and consent was not necessary.
Cell lines and plasmids
The Gal-32 cell line was generously provided by Dr. EH Chu (1). Antibiotics were supplied by BRL (Bethesda, MD).All restriction enzymes were provided by New England Biolabs, Ipswich, MA. Cells were cultured at 37°C in the presence of 5% CO2. Complete growth medium is GIBCO α-minimal essential medium (GIBCO, now Life Technologies, Bethesda, MD) containing 292 mg/L of glutamine, 17.9 mM NaHCO3, 8.4 mMNaCl, 3-5% heat-inactivated fetal calf serum, 37μM hypoxanthine (H), 21 μM thymidine (T), and 22 mM D-glucose. Galactose medium is complete medium with 22 Mm galactose in place of glucose and 3-5% dialysed fetal calf serum (BRL, now Life Technologies, Bethesda, MD) (unless otherwise noted) in place of undialysed serum. MXHAT (BRL, now Life Technologies, Bethesda, MD) medium is complete medium containing 78 mM mycophenolic acid (M), 1.6 mM xanthine (X), 0.1 mM hypoxanthine, 4.5 μM aminopterin (A), 40 μM thymidine and no uridine[13]. G418 (BRL, now Life Technologies, Bethesda, MD) medium is complete medium plus 400 or 800 μg/ml G418 (GIBCO, ~50% pure) [14].
The human cosmid library and the pSV13 cosmid vector in Eschrechia coli Hb101 were generously donated by Dr. Mary McCormick [15]. pSV13 contains genes for chloramphenicol resistance (camr) and xanthine-guanine phosphoribosyl-transferase (SV2-gpt); the human (HeLa) genomic DNA (average size 40 kb) was inserted into the single PstI site of pSV13. The pSV2-neo plasmid containing the gene that confers resistance to the neomycin analog G418 was constructed by Drs. P. J. Southern and P. Berg. The cosmid vector pCV108, containing the ampr and SV-neo genes, was generously supplied by Dr, Y.-F. Lau [16]. The Blur-8 clone, constructed by Deininger et al. [17], was kindly donated by Dr. Antonio Fojo. E. coli strain Sure-1 {mcrA, Δ[mcrCBhsdSMR- mrr] 171, sbcC, recB, recJ, umuC::Tn5 (kanr), uvrC, supE44, lac, gyrA96, relA1, thi-1, endA1 [F’ proAB, laclqZΔM15, Tn10, (tetr]} was obtained from Stratagene (La Jolla, CA).
Gene transfer
Gene transfer was performed using slight modifications of the CaPO4-DNA (Sigma, St. Louis, MO) precipitation methods of Abraham et al. and Chen and Okayama [18,19, 20]. Gal-32 cells, at density of 5 x 105 / 100 mm dish, were incubated overnight in 10 ml of complete medium, then exposed to a mixture of CaPO4-DNA for 6.5 h (Table 1) or 15 h (Table 3) with 5% CO2(Table 1, experiments 1 and 2)or 3% CO2(Table 1, experiment 3; Table 3). The CaPO4-DNA precipitate was prepared with 10 μg/100 mm dish (Table 1) or 20 μg/100 mm dish (Table 3) of each purified DNA and 4-(2-Hydroxyethyl)-1- piperazineethanesulfonic acid (Hepes, BRL, Bethesda, MD) pH 7.12 (17) (Table 1) (experiments 1 and 2), Hepes, pH 6.95 [19,20](Table 1, experiment 3), or N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid (BES), pH 7.0 (Table 3). After washing the cells with complete medium (Table 1) or phosphate-buffered saline (Table 3), the cells were incubated in complete medium for 40 h (Table 1, experiments 1 and 2), 72 h (Table 1, experiment 3), or 24 h (Table 3) and then re-plated.
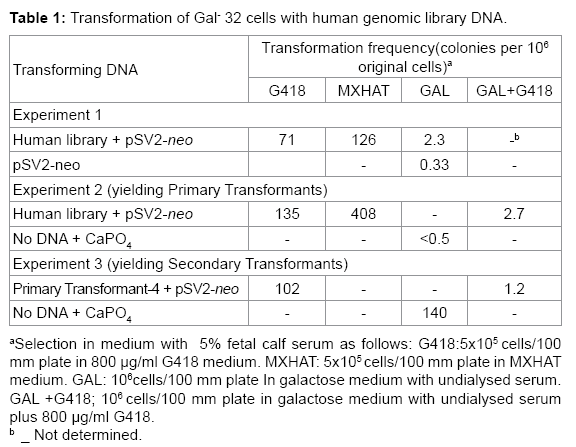
Table 1: Transformation of Gal- 32 cells with human genomic library DNA.
DNA purification
For large plasmid purifications, bacteria containing plasmids were grown in Super Broth for 24 – 48 h or in Luria-Bertani medium (LB) plus antibiotic and amplified with chloramphenicol [21]. Plasmids purified with Qiagen (Germany) columns were grown in Terrific Broth. Plasmid DNA used for gene transfer, for probes, and for construction of the primary transformant library were purified either by double banding in a CsCl-ethidium bromide density gradient or with Qiagen 500 columns. The human genomic cosmid library in host E. coli HB101 was grown in Super Broth plus cholrampheicol at 30 μg/ml for 24 h and purified by double banding in a CaCl-ethidium bromide density gradient [22].
For plasmids mini preperations, the alkaline lysis method was used to purify pCV108-transformant recombinant cosmid DNA from camrSure-1 bacteria, grown in LB plus chloramphenicol at 30 μg/ml [21].
Primary transformant genomic DNA that was used for gene transfer to yield secondary transformants was purified according to the method of Abraham et al.
The human Alu probe was isolated by digestion of the purified Blur-8 plasmid DNA with BamHI, agarose gel electrophoresis, and electroelution of the 300 bp insert from the gel [21]. Similarly, the 560 bpcamr probe was isolated following digestion of the purified pSV14 plasmid DNA with EcoRI and SalI [21]. The probes were labeled with [α-32P]dCTP (DuPont) using a nick translation kit or random primer kit (Bethesda Research Laboratories (BRL), Bethesda, MD).
Southern blotting and hybridization
DNA digestion with restriction endonucleases was separated by 0.8% agarose gel electrophoresis in 89 mMTris borate-2 mM EDTA buffer and blotted to GeneScreen membranes (New England Nuclear Corp., Boston, MA) [23]. The Gene Screen blot was hybridized in 50% formamide at 42°C with the 32P-labeled probes as previously described [23].
To eliminate background with the genomic DNA blots, the heatdenatured (95°C, 5 min) radioactive probe was prehybridized to a sheet of blank GeneScreen for 2-3 h in the hybridization solution. The prehybridized probe was denatured at 80°C for 10 min immediately before addition to the DNA blot. This step was omitted for hybridization to the pCV108-transformant recombinant plasmid DNA.
After hybridization for 16-24 h, the membrane was washed with constant agitation as follows: 1) three times with 330 ml of 2X SSPE (0.36 M NaCl, 20 mMNaPi, pH 7.4, 2 mM EDTA) and 0.1% SDS (Sigma, St. Louis, MO) at room temperature for 10 min; 2) twice with 250 ml of the 2X SSPE and 1% SDS at 60°C for 20 min; and 3) three times with 330 ml solution 0.1X SSPE and 0.1% SDS for 20 min at 52°C for the Alu probe and at 65°C for the camr probe. The blots were exposed to Kodak XRP-5 film (Kodak, NY) at -70°C until the bands were visible. The probe was removed by incubating the GeneScreen membrane in a final solution of 0.02 M Tris-HCl, (Sigma) pH 7.0, 0.01 M EDTA and 96% (v/v) formamide (Fisher Scientific, Hanover Park, IL) for 1 h at 80°C.
Construction of a Gal+ transformant genomic DNA library
A genomic library was constructed from primary Gal+transformant-3 (1-TR-3) in the cosmid cloning vector, pCV108. The library was screened by replica-plating on plates containing chloramphenicol.
Preparation and partial digestion of high molecular weight genomic DNA
For each genomic DNA preparation, seven T-150 cm2 tissue culture flasks were employed to grow 7.0 x 107 1-TR-3 cells in galactose and MXHAT media. High molecular weight genomic DNA was purified by CsCl-ethidium bromide (density gradient centrifugation following the protocol of Fleischmann et al. The size of the DNA was analyzed by electrophoresis in a 0.3% (v/w) agarose gel using oligomers of bacteriophage lambda DNA as markers (BRL, Bethesda, MD). The purified high molecular weight genomic DNA (average size, 150 kb) was partially digested with the restriction enzyme MboI that recognizes a 4-bp sequence. This partial digestion yields overlapping fragments and cohesive ends which anneal with the BamHI digested pCV108 vector. Analytical pilot experiments were performed varying the time of MboI digestion. The time of digestion that gave the brightest staining band of DNA between 23 and 50 kb was determined and half of this time was then used in the preparative digestion procedure as described by Seed et al. [24]. Therefore, 40 μg of genomic DNA was digested with20U of MboI for 1.0 min at 37°C. The resulting fragments were phenolextracted, ethanol-precipitated, and size-fractioned by 10- 40% sucrose density gradient centrifugation. Following analytical electrophoresis in a 0.3% agarose gel, 35-45 kb gradient fractions were pooled and used for construction of the primary transformant cosmid library.
Digestion and dephosphorylation of vector DNA
The pCV-108 cosmid vector containing the SV2-neo and ampr genes was used to construct the primary transformant genomic DNA library. Purified pCV108 DNA was digested to completion with BamHI, phenol-extracted, and ethanol-precipitated. The linearized vector DNA was dephosphorylated with calf intestinal alkaline phosphatase[16,21].
DNA ligation
In order to determine an optimal set of conditions for ligation between vector and genomic DNA, a series of test ligation reactions at different concentrations of DNA were performed. The optimal results were obtained with a molar ratio of 9:1 (weight ratio of 2:1) of vector DNA (8.9 kb, BamHI/phosophatase-treated pCV108) to genomic DNA (35-45 kb, MboI/sucrose-fractioned 1-TR-3). The ligation of the DNA fragments possessing cohesive ends was accomplished using the procedure of DiLella and Woo except that polyethene glycol was omitted from the ligation buffer. The reactions were incubated at 14°C for 14-16 h and the products were analyzed by electrophoresis in a 0.2% agarose gel [25].
In vitro packaging
In vitro packaging of the ligated recombinant DNA or the vector DNA (control reaction) was performed with bacteriophage λ packaging kits from Stratagene or Bethesda Research Laboratories according to the specifications of the manufacturers with a slight but critical modification: The DNA was added to a mixture of the two packaging extracts by thawing the freeze-thaw-lysate (10μl), immediately adding it to the frozen sonicated extract (15 μl), within 5-7 s pipetting in the ligated DNA (vector + genomic, 595 ng in 2.66 μl), and quickly stirring the packaging reaction with the tip of the pipette. The packaging mixture was immediately centrifuged for 5 s and incubated at 22°C for 2 h. The immediate addition of the recombinant DNA to the extracts and the temperature of incubation are very crucial to the success of the packaging reaction.
Transduction of packaged DNA and screening the library
After tittering, the library was plated at high density on LB plates containing ampicillin and screened by replica plating on LB plates containing chloramphenicol.
For transduction, Sure-1 bacteria (Stratagene, La Jolla, CA) were grown in LB medium containing 0.4% maltose and 10mM MgCl2 at an OD600 of 0.5 (Stratagene procedure). The cosmid library was tittered by mixing an aliquot of the packaging reaction with the Sure- 1 bacteria and spreading on LB agar plus ampicillin (50 μg/ml) plates (150 mm) that were overlaid with detergent-free nitrocellulose filters (Millipore, HATF) [21]. For the large scale transduction, a mixture of the packaging reaction and Sure-1 bacteria were spread at high density (50,000 colonies/150 mm dish) on twenty LB plus ampicillin plates overlaid with nitrocellulose filters. The plates were incubated at 37°C until tiny (0.1-0.2 mm) colonies appeared in 8 to 12 h. The colonies on the LB plates containing ampicillin were replicated onto LB plus chloramphenicol (30 μg/ml) or ampicillin plates that were overlaid with nitrocellulose filters [21,26].
The master filter was stored on LB agar plates containing ampicillin and 25% glycerol at -70°C. The remainder of the packaging reaction containing the cosmid DNA library was stored in 7% (v/v) dimethyl sulfoxide at -70°C. Before further analysis, the camr recombinant colonies were purified by spreading on LB plates containing chloramphenicol and picking single colonies.
Transfer of the human wild-type Gal+32 gene into Gal-32 cells
The objective of this study was to clone the wild type human gene that complements the Gal-32 mutation. Gal-32 cells were cotransformed with pSV2-neo plasmid DNA [13] and recombinant DNA from a human genomic library [15] containing the dominant human Gal+ gene. The pSV13 cosmid vector used in construction of the human library carries the SV2-gpt gene, which permits mammalian cells to grow in MXHAT [13], as well as camr gene, which enables bacterial cells to grow in chloramphenicol. The dominant SV2-neo gene allows mammalian cells to grow in the neomycin analog G418. The frequency of transfer of the SV2-neo gene (7.1-13.5 in 105 cells in G418) and the SV2-gpt gene (1.26-4.08 in 104 cells in MXHAT) was high, as expected for introduction of a cloned gene (Table 1). In experiment 1, Table 1, Gal-32 yielded spontaneous CHL Gal+ revertants with pSV2-neo at the usual frequency of 0.33 in 106 cells. However, Gal+ transformants with the human genomic library appeared at a 7-fold higher frequency than spontaneous revertants (2.3 compared to 0.33 in 106 cells in galactose, Table 1, experiment 1).
Primary Gal+ transformants were isolated by transferring human genomic library DNA and pSV2-neo DNA into Gal-32 cells and selecting for growth in galactose plus G418. The frequency of isolation of primary transformants (2.7 x 10-6, Table 1, experiment 2) was 105 times higher than that (0.5 x 10-6 multiplied by 135 x 10-6) expected for a CHL Gal+ revertant that incorporated the SV2-neo gene.
Secondary transformants were isolated by transferring primary transformant-4 DNA and pSV2-neoDNA into Gal-32 cells and selecting for growth in galactose plus G418. Although some CHL Gal+ revertants may have also been present, the frequency (1.2 x 10-6, Table 1, experiment 3) of isolation of secondary Gal+ transformants in galactose and G418 was 100 times higher than that (102 x 10-6 multiplied by 140 x 10-6) expected for a CHL Gal+ revertant that incorporated the SV2-neo gene.
Plating efficiency of transformants
Isolated colonies of primary transformants exhibited high relative plating efficiency (77-100%) in galactose or G418 and low relative plating efficiency (7-9%) in MXHAT (Table 2). Survival in MXHAT indicates that the primary transformants contain the unselected SV2- gpt gene, which is located in the cosmid vector used to construct the human genomic library. Isolated colonies of secondary transformants also demonstrated high relative plating efficiency (79-100%) in galactose or G418. About half of the secondary transformants initially grew in MXHAT, but did not grow in MXHAT after expansion and further testing (Table 2). The human Gal+ was based on (1, growth in MXHAT, which was unselected, and (2, the frequency of isolation of primary and secondary transformants growing in galactose + G418 compared to that expected for CHL Gal+ revertants growing in these media.
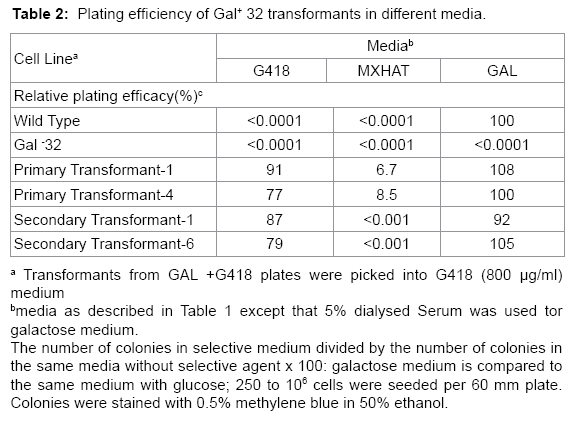
Table 2: Plating efficiency of Gal+ 32 transformants in different media.
Detection of human Alu and bacterial camr DNA sequences in the primary and secondary transformants
camr and human Alu probes (Figure 1). The camr gene is present in the pSV13 vector used to construct the human genomic library [15]and may still be linked to the human Gal+ gene in the transformants. The highly repeated human Alu sequences, which occur every 2-3 kb in the human genome [27], were also expected to be linked to the human Gal+ gene.
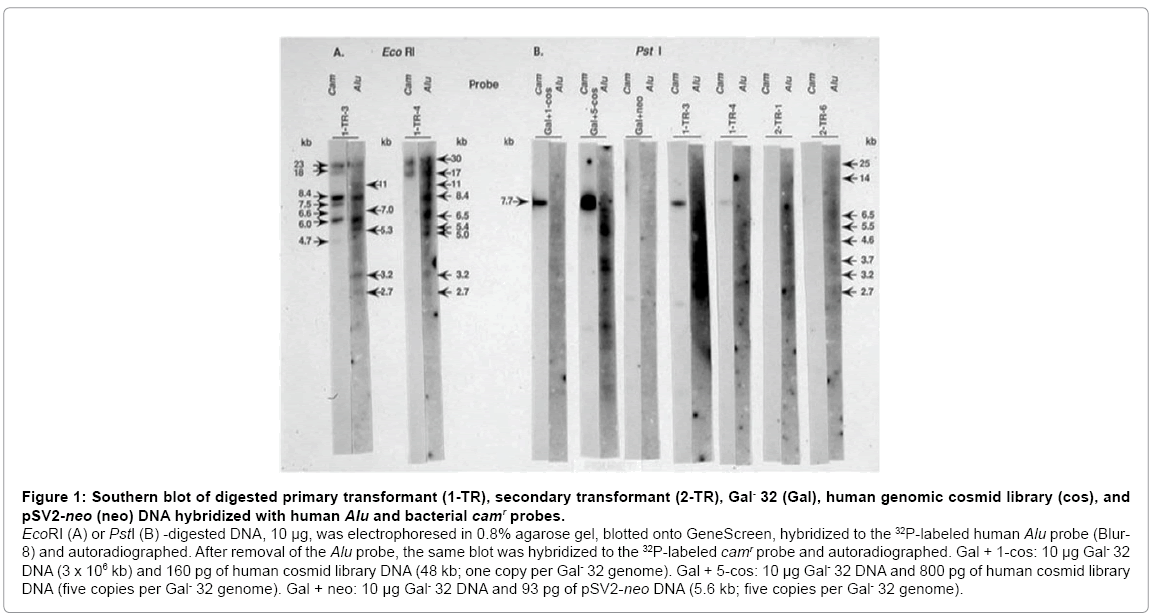
Figure 1: Southern blot of digested primary transformant (1-TR), secondary transformant (2-TR), Gal- 32 (Gal), human genomic cosmid library (cos), and pSV2-neo (neo) DNA hybridized with human Alu and bacterial camr probes.
EcoRI (A) or PstI (B) -digested DNA, 10 μg, was electrophoresed in 0.8% agarose gel, blotted onto GeneScreen, hybridized to the 32P-labeled human Alu probe (Blur- 8) and autoradiographed. After removal of the Alu probe, the same blot was hybridized to the 32P-labeled camr probe and autoradiographed. Gal + 1-cos: 10 μg Gal- 32 DNA (3 x 106 kb) and 160 pg of human cosmid library DNA (48 kb; one copy per Gal- 32 genome). Gal + 5-cos: 10 μg Gal- 32 DNA and 800 pg of human cosmid library DNA (five copies per Gal- 32 genome). Gal + neo: 10 μg Gal- 32 DNA and 93 pg of pSV2-neo DNA (5.6 kb; five copies per Gal- 32 genome).
The Gal+ transformants hybridized to both the human Alu probe and the bacterial camr probe (Figure 1). As expected, both probes hybridized to the human cosmid library (Figure 1B, Gal+ 1-cos, Gal+ 5-cos) and did not hybridize to the CHL DNA nor the pSV2-neo DNA (Figure 1B, Gal+ neo). The primary transformants (1-TR-3 and 1-TR-4) and the secondary transformants (2-TR-1 and 2-TR-6) hybridized to the human Alu probe, indicating the presence of human DNA. It is of interest that the primary transformants, 1-TR-3 and 1-TR-4, have the following Alu-hybridizing, EcoRI fragments in common: 11 kb, 8.4 kb, 5.3 kb, 3.2 kb, and 2.7 kb.
The primary transformants also hybridized to the camr probe, demonstrating the presence of the pSV13 vector. During gene transfer, a large concatemer of unlinked DNA is formed and the integrated into the chromosomal DNA [18]. Since the camr probe is an EcoRISalI fragment derived from pSV13, each integrated cosmid copy was expected to produce a single band after hybridization of this probe with EcoRI-digested genomic DNA from the transformants. With the camr probe seven bands were observed with EcoRI-digested 1-TR-4 (Figure 1A). Furthermore, in the EcoRI-digested primary transformants, multiple Alu-hybridizing bands which have the same mobility as camrhybridizing bands (23 kb, 8.4 kb, 6.0 kb, in 1-TR-3; 30 kb, 17 kb in 1-TR-4) were observed.
PstI digestion of the human cosmid library or the primary transformants yielded a 7.7 kb fragment that hybridized to the camr probe (Figure 1B, Gal+ cos, 1-TR-3, 1-TR-4). This fragment corresponds to the linear, intact pSV13 vector since the human genomic DNA was inserted into the unique PstI site of the pSV13 vector. The intensities of the 7.7 kb camr bands suggests that there is approximately one copy of the pSV13 vector (Gal+cos) per genome in 1-TR-3 and much less than one copy in 1-TR-4. Even though the camr probe did not detect any bands in PstI-digested secondary transformants (Figure 1B, 2-TR-1, 2-TR-6), in another experiment not shown, a camr band was observed in EcoRI-digested secondary transformant-1.
Mitochondrial protein synthesis in transformants
To verify that the defect in mitochondrial protein synthesis was corrected in the putative Gal+transformants, cells were labeled with [35S]-methionine in the presence of a cytoplasmic protein synthesis inhibitor (cycloheximide), followed by the isolation of mitochondria and analysis of the resulting labeled proteins on SDS polyacrylamide gels (Figure 2). The absence of all labeled proteins (Figure 2, 1-TR-4 + CAP) in the presence of cycloheximide and chloramphenicol, a specific inhibitor of mt protein synthesis, indicates that the proteins labeled in the presence of cycloheximide are indeed mitochondrially synthesized. The intensity of the bands and the mobility pattern of the proteins in the primary and secondary transformants were similar to those of the wildtype whereas a significant reduction in the level of the same proteins (except ATPase 6) was observed in Gal-32 (Figure 2). These results demonstrate that all the mitochondrially synthesized proteins that were reduced in the mutant are restored to normal wild-type levels in both primary and secondary transformants. Considering the gene transfer, hybridization, and mt protein synthesis data, we conclude that a human gene is present in both the primary and secondary transformants and is capable of correcting the Chinese hamster Gal- 32 mutation.
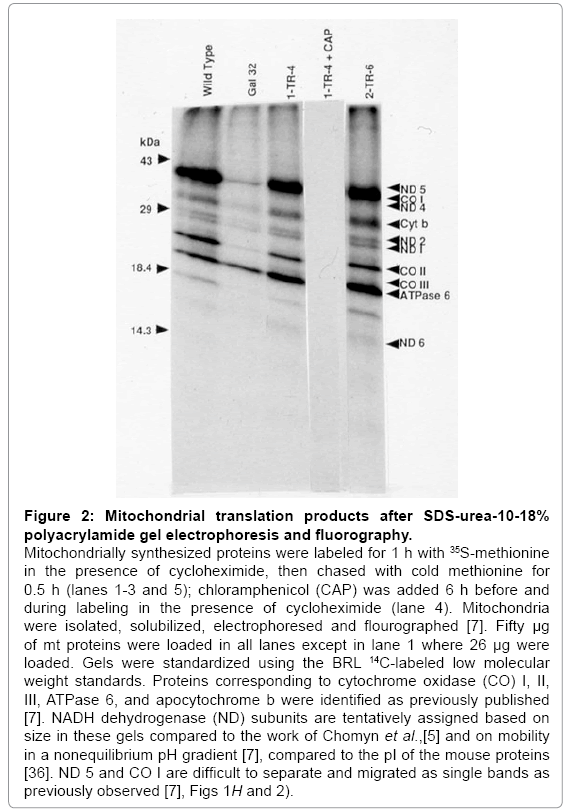
Figure 2: Mitochondrial translation products after SDS-urea-10-18% polyacrylamide gel electrophoresis and fluorography.
Mitochondrially synthesized proteins were labeled for 1 h with 35S-methionine in the presence of cycloheximide, then chased with cold methionine for 0.5 h (lanes 1-3 and 5); chloramphenicol (CAP) was added 6 h before and during labeling in the presence of cycloheximide (lane 4). Mitochondria were isolated, solubilized, electrophoresed and flourographed [7]. Fifty μg of mt proteins were loaded in all lanes except in lane 1 where 26 μg were loaded. Gels were standardized using the BRL 14C-labeled low molecular weight standards. Proteins corresponding to cytochrome oxidase (CO) I, II, III, ATPase 6, and apocytochrome b were identified as previously published [7]. NADH dehydrogenase (ND) subunits are tentatively assigned based on size in these gels compared to the work of Chomyn et al.,[5] and on mobility in a nonequilibrium pH gradient [7], compared to the pI of the mouse proteins [36]. ND 5 and CO I are difficult to separate and migrated as single bands as previously observed [7], Figs 1H and 2).
Construction of a primary transformant cosmid library
Since primary transformant-3 exhibits multiple EcoRI restriction fragments that hybridize to both the human Alu probe and to the camr probe (Figure 1), it is likely that the human DNA is still linked to the camr gene as in the pSV13-human cosmid DNA library used to obtain the primary transformant. Therefore, a genomic DNA library was constructed from primary Gal+ transformant-3 and the cosmid cloning vector, pCV108. High molecular weight DNA (>150 kb) was purified from primary transformant-3 and partially digested with MboI. MboI genomic DNA fragmetns of 35–50 kb were isolated from a sucrose density gradient. The pCV108 cosmid vector was completely digested with BamHI to linearize it and to produce ends complementary to the MboI-digested genomic DNA. The digested vector was treated with alkaline phosphatase to remove the 5’-terminal phosphate and thereby suppress self-ligation. None of the phosphatase-treated, BamHIdigested pCV108 changed mobility after incubation with T4 DNA ligase, indicating the absence of self-ligation (Figure 3, pCV108 + ligase). When the MboI-digested, size fractioned, primary transformant-3 DNA was ligated to the BamHI-digested, dephosphorylated pCV108 vector, all of the transformant DNA was converted to a larger size DNA (Figure 3, pCV108 + 1-TR-3 ± ligase). Since there was a nine to one molar excess of vector DNA to genomic DNA, most of the vector DNA did not yield a larger size DNA. The recombinant DNA was packaged into bacteriophage λ particles, and transduced into E. coli.
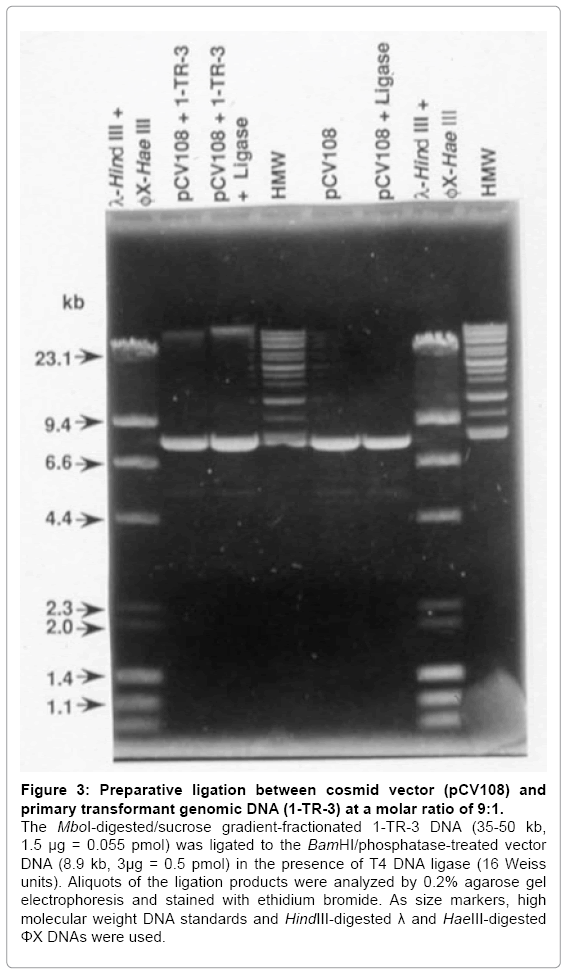
Figure 3: Preparative ligation between cosmid vector (pCV108) and primary transformant genomic DNA (1-TR-3) at a molar ratio of 9:1.
The MboI-digested/sucrose gradient-fractionated 1-TR-3 DNA (35-50 kb, 1.5 μg = 0.055 pmol) was ligated to the BamHI/phosphatase-treated vector DNA (8.9 kb, 3μg = 0.5 pmol) in the presence of T4 DNA ligase (16 Weiss units). Aliquots of the ligation products were analyzed by 0.2% agarose gel electrophoresis and stained with ethidium bromide. As size markers, high molecular weight DNA standards and HindIII-digested λ and HaeIII-digested ΦX DNAs were used.
The primary transformantcosmid library was plated on ampicillin to select for the presence of the pCV108 vector, which contains the ampr gene. The library was replica-plated onto chloramphenicol because the camr gene is present in the primary transformant and is likely to remain linked to the human DNA. The total library obtained was 1.9 x 106ampr colonies. Only half of these colonies were replica-plated on chloramphenicol since this number would be more than enough for a 99% probability of isolating a single copy gene [14]. There was an average of 7 camr clones per 50,000 ampr colonies isolated. The total number of camr clones recovered was 144 from 9.5 x 105ampr colonies. Based on the average size of the insert (3 x 104 base pairs) and the mammalian genome size (3 x 109base pairs), this corresponds to approximately 10 copies of the camr gene per genome. The average size of the DNA inserts from 32 camr recombinant clones was 35 kb.
These camr colonies were digested with EcoRI, electrophoresed, transferred to GeneScreen, and hybridized to a 32P-labeled Alu probe. Of the 32 pCV108-transformant recombinant plasmids digested with EcoRI, all exhibited some DNA fragments of approximately the same size that were not vector fragments. After Southern blotting, 28 of these 32 plasmids hybridized to a human Alu probe at a location or intensity different from that of the vector; 10 of these exhibited very distinct Aluhybridizing signals. Figure 4 shows eight of these camr recombinant clones. The Alu probe also hybridized very weakly with the EcoRIreleased cosmid vector (~8.9 kb). Since the vector was released intact (as evidenced by its size) by EcoRI digestion, its weak hybridization with the Aluprobe may be attributed either to the presence of short Alulike sequences within its DNA structure or contamination of the probe with pBR vector sequences.
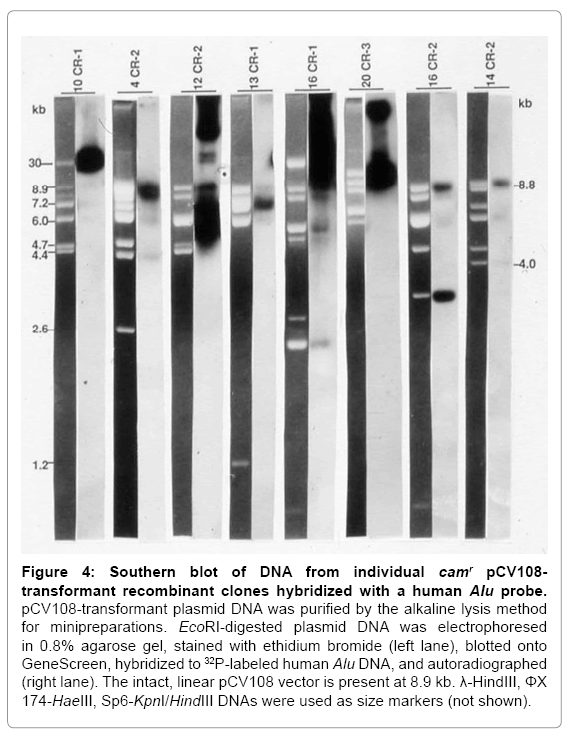
Figure 4: Southern blot of DNA from individual camr pCV108- transformant recombinant clones hybridized with a human Alu probe.
pCV108-transformant plasmid DNA was purified by the alkaline lysis method for minipreparations. EcoRI-digested plasmid DNA was electrophoresed in 0.8% agarose gel, stained with ethidium bromide (left lane), blotted onto GeneScreen, hybridized to 32P-labeled human Alu DNA, and autoradiographed (right lane). The intact, linear pCV108 vector is present at 8.9 kb. λ-HindIII, ΦX 174-HaeIII, Sp6-KpnI/HindIII DNAs were used as size markers (not shown).
Gal-32 mutation is complemented with DNA from pCV108- transformant recombinant clones
The identification of the recombinant clone that corrects the Gal- 32 mutation ultimately depends on gene transfer since the product of the gene is not known. To identify which one of the Alu-hybridizing, camr recombinant cosmid clones contains the Gal+ 32 gene, purified DNA from each recombinant clone was used to transform Gal-32 mutant cells selecting for growth in GAL or Gal+ G418. Even though the efficiency of transformation was high with all eight clones for the SV2-neo gene (G418 selection), only two of the recombinant clones, 10CR-1 and 4CR-2, enabled the mutant to grow in GAL or Gal+ G418 (Table 3). Eight and four colonies per 106 cells were recovered in GAL with 10CR-1 and 4CR-2, respectively. Similarly, in the double selection (Gal+ G418) three times more colonies were retrieved with 10CR-1 than with 4CR-2.
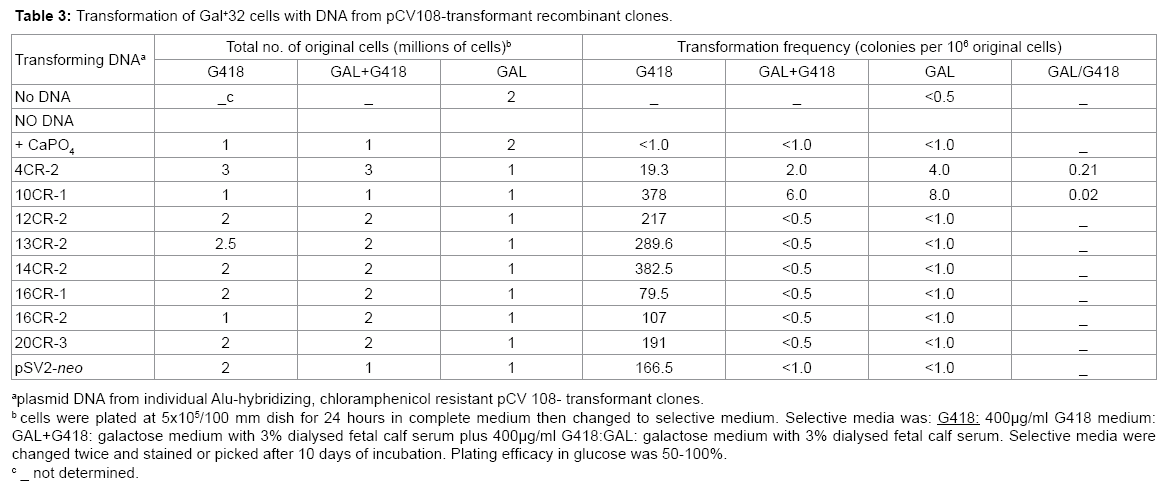
Table 3: Transformation of Gal+32 cells with DNA from pCV108-transformant recombinant clones.
In case that expression of the Gal+ gene was extremely low, the G418-resistant transformant colonies for each cosmid clone were also pooled, grown in complete medium, and plated at different cell densities in GAL or GAL and G418 media. As a negative control for the presence of human DNA inserts, the pSV2-neo-transformants were cultured as well. The same results were obtained as in Table 3; only 4CR- 2 and 10CR-1 produced transformants in GAL and Gal+ G418 (data not shown).
Mitochondrial protein synthesis is restored in Gal-32 complementing recombinant clones
To determine if the putative Gal+ transformants restored mitochondrial protein synthesis, growing cells were labeled with [35S] methionine in the presence of cycloheximide and proteins from isolated mitochondria were analyzed on SDS-urea polyacrylamide gels. In Gal- 32 cells transformed with 10CR-1, mitochondrial protein synthesis is restored to the wild-type level; while mitochondrial protein synthesis is greatly reduced in the mutant (Figure 5). When expanded to test for mitochondrial protein synthesis, the Gal+ phenotype was not stable in Gal-32 cells transformed with 4CR-2; therefore, mitochondrial protein synthesis could not be accurately measured in this case.
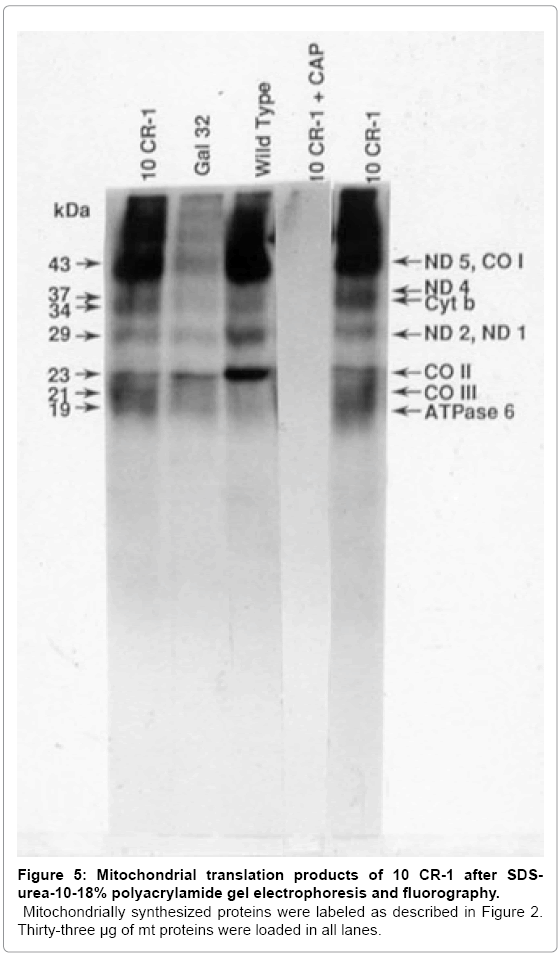
Figure 5: Mitochondrial translation products of 10 CR-1 after SDSurea- 10-18% polyacrylamide gel electrophoresis and fluorography.
MitochondriaIly synthesized proteins were labeled as described in Figure 2. Thirty-three μg of mt proteins were loaded in all lanes.
Restriction digestion of 10CR-1 and 4CR-2
Since both 10CR-1 and 4CR-2 complemented the Gal-32 mutation, purified DNA from these clones was digested with different restriction enzymes (Figure 6). When 10CR-1 was digested with EcoRI, four fragments were observed: 7 kb (vector), 5.7 kb, 4.7 kb, and 4.6 kb; therefore, the vector size (7 kb) and total insert size (15 kb) are smaller than expected (8.0 kb and 35 kb, respectively). Cosmid clones are known to rearrange. Interestingly, digestion of 10CR-1 with BamHI, BssHII, PvuI, or SmaIall resulted in three fragments with nearly the same size (11.7 kb, 5.8 kb, 4.5 kb for BamHI, Figure 6). Double digestions revealed that the sequence BssHII, SmaI, BamHI, EcoRI, PvuIis repeated three times (Figure 7). Two of these BssHII-SmaIBamHI- EcoRI-PvuI sequences (a and c sites) do not hybridize to the Aluprobe; while the cluster with EcoRI-PvuIin the vector (d sites) does hybridize to the Aluprobe.
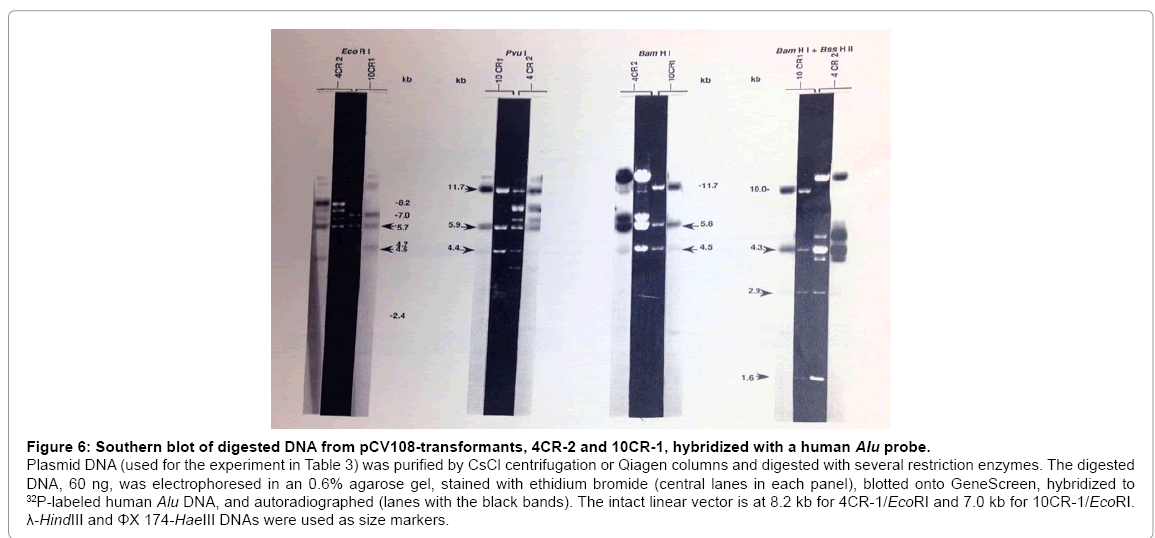
Figure 6: Southern blot of digested DNA from pCV108-transformants, 4CR-2 and 10CR-1, hybridized with a human Alu probe.
Plasmid DNA (used for the experiment in Table 3) was purified by CsCl centrifugation or Qiagen columns and digested with several restriction enzymes. The digested DNA, 60 ng, was electrophoresed in an 0.6% agarose gel, stained with ethidium bromide (central lanes in each panel), blotted onto GeneScreen, hybridized to 32P-labeled human Alu DNA, and autoradiographed (lanes with the black bands). The intact linear vector is at 8.2 kb for 4CR-1/EcoRI and 7.0 kb for 10CR-1/EcoRI. λ-HindIII and ΦX 174-HaeIII DNAs were used as size markers.

Figure 7: Restriction map and Alu hybridization of pCV108-recombinant clone 10CR-1 and restriction map of pCV108.
Clone 10CR-1 was digested with EcoRI, BamHI, SmaI, BssHII, ClaI, PvuI, HindIII, KpnI, EcoRI + BamHI, ClaI + PvuI , BamHI +SmaI, BamHI + BssHII, SmaI + BssHII, BssHII + ClaI, SmaI + ClaI, SmaI + KpnI, BssHII + KpnI, BamHI + KpnI, BamHI + ClaI. The pCV108 vector was digested with HpaI, SmaI, BssHII, ClaI, PvuI, HindIII, KpnI (no digestion), SalI, HpaI + ClaI, HpaI + SmaI, BssHII + HindIII, HpaI + BssHII, SmaI + HindIII, PvuI +HindIII, ClaI + SalI. Electrophoresis, blotting, and hybridization with the human Alu probe are described in Figure 6. The striped regions of 10CR-1 hybridized to the Alu probe. The solid region of 10CR-1 is the altered pCV108 vector. The pCV108 map is based on this data and published maps of the component parts [19].
EcoRI digestion of 4CR-2 resulted in the expected ~8.2 kb vector and several insert fragments with a total size of ~36 kb. With 4CR-2 and 10CR-1, several restriction fragments with the same size and same Alu-hybridizing properties were observed (Figure 6). In 4CR-2 and 10CR-1, there are 5.7 kb EcoRI, 5.9 kb PvuI, 5.8 kb BamHI, and 4.3 kb BamHI/BssHII fragments which hybridize to the Alu probe. In addition, both 4CR-2 and 10CR-1 contain 4.6 kb EcoRI, 4.4 kb PvuI, 4.5 kb BamHI, 2.9kb BamHI/BssHII, and 1.6kb BamHI/BssHII fragments which do not hybridize to the Alu probe. The fragments which do not hybridize to the Aluprobe could represent human Gal+ DNA or CHL DNA. Fragments with the same size and Alu-hybridizing properties in 4CR-2 and 10CR-1 are likely to encode the same portion of the human Gal+ gene.
Gal-32 is a Chinese hamster lung cell mutant that is unable to grow in galactose or fructose due to a differential decrease in mitochondrially synthesized proteins [7]. In a separately published data, it was demonstrated that the majority of the Rhodamine 6-G-treated hybrids grew in galactose as expected for a nuclearly encoded gene considering that Rhodmnine 6-G interferes with transmission of mt DNA but not nuclear DNA. Therefore, these results are compelling in their demonstration of the nuclear origin of the Gal-32 mutation [28].
The objective of this research was to isolate a human gene that complements the Gal-32 mutation. Recessive Gal-32 cells were cotransformed with pSV2-neo plasmid DNA and recombinant DNA from a human genomic library containing the dominant human Gal+ gene and a camrgene present in the pSV13 vector (14). Primary transformants were selected by growth in GAL and G418. The double selection in Gal+ G418 was utilized to distinguish cells that were Gal+ as a result of incorporating the human gene from cells that were Gal+ because of the spontaneous reversion of the CHL Gal-32 gene. Hybridization of primary transformant DNA with a human Alusequence and a camr probe confirmed the presence of human DNA sequences that may remain linked to the camrgene. In order to rescue the human Gal+ gene, a genomic library was constructed with primary transformant-3 DNA and the pCV108 cosmid vector, which contains ampr and SV2-neo genes. The pCV108-transformant library was plated on ampicillin and replica plated onto chloramphenicol. Thus, the camr gene was used to identify clones with the nearby human sequences. In fact, most of the camr clones contained human sequences that hybridized to the human Alu probe. DNA from two camr, Alu-hybridizing clones (10CRA and 4CR-2) was able to transform recessive Gal-32 cells to the Gal+ phenotype and, in the case of 10CR-1, to restore mitochondrial protein synthesis.
Primary and secondary transfortnants
During gene transfer, 1- 6 copies of the plasmid are integrated into high molecular weight DNA in tandem or at separate sites [14, 19]. The camr probe hybridized to seven different EcoRI-primary transformant-3 (1-TR-3) restriction fragments, suggesting that pSV13 -camr-human cosmids had integrated at seven different sites (Figure 1). Hybridization of the camr probe to the PstI-digested 1-TR-3 DNA indicated that there is approximately one pSV13 cosmid copy per genome. On the other hand, in the pCV108-transformant library, the frequency of camr colonies among the ampr colonies indicates that there are 10 copies of the pSV13-camr-human cosmids per genome. The explanation for these contradictory pSV13 cosmid copy numbers in 1-TR-3 is not clear.
The frequency of gene transfer with a single copy gene using genomic DNA is expected to be at least 200 times lower, than the frequency with a cloned gene [18, 29]. In this study (Table 1), the transformation frequency with the cloned SV2-neo (G418 selection, 135 or 102 in 106 cells) or SV2-gpt(MXHAT selection, 126 or 408 in 106 cells) genes was at the expected level [13]. Surprisingly, the frequency of isolation of primary transformants (GAL and G418, 2.7 in 106 cells) and secondary transformants (GAL and G418, 1.2 in 106 cells) was higher than expected: only 50-150 times lower than the frequency for cloned SV2-neo or SV2-gptgenes. The unexpectedly high frequency of isolation of primary transformants may be due to multiple copies of the Gal+ gene in the human cosmid library, which may have arisen during amplification of the library [30]. Gene amplification is also known to occur during gene transfer, perhaps accounting for the high frequency in isolating secondary transformants.
Human gene complements the Gal-32 mutation
We conclude that a human gene was transferred into Gal-32 cells and corrected the Chinese hamster mutation for the following reasons: 1) The frequency of isolation of primary Gal+ transformants in Gal+ G418 media was 105 times higher than expected for a CHL Gal+ revertant that incorporated the SV2-neo gene; 2) the frequency of isolation of secondary Gal+ transformants in Gal+ G418 media was 100 times higher than expected for a CHL Gal+revertant that incorporated the SV2-neo gene; 3) primary transformants were able to grow in MXHAT media even though the SV2-gpt gene present in the pSV13 vector was unselected; 4) detection of multiple human Aluhybridizing DNA in primary and secondary transformants; 5) detection of multiple camr-hybridizing restriction fragments corresponding in size to the Alu-hybridizing fragments indicated the presence of multiple human-pSV13 recombinant clones in the primary transformants; 6) the restoration of mt protein synthesis in primary and secondary transformants. These data also support the conclusion that the Gal-32 gene is nuclearly encoded, since mtDNA has not been reported to yield stable transformants by CaPO4 precipitation. It is significant that the human gene corrects the Chinese hamster Gal-32 mutation because many human mt proteins are unable to substitute for Chinese hamster proteins. For example, in human-rodent or mouse-rat hybrid cell lines, mtDNA is usually not maintained unless nuclear DNA from the same species is also retained [31, 32].
Isolation of two recombinant human clones that correct the Gal-32 mutation
Two pCV108-transformant clones (10CR-1 and 4CR-2) that complement the Gal-32 mutation were identified by transferring the purified recombinant cosmid DNA into Gal-32 cells and selecting for growth in Gal+ G418. If a particular pCV108-recombinant clone contains the Gal+ gene, the transformation frequency in galactose is expected to be about the same as the transformation frequency in G418. However, when plasmid DNA from 4CR-2 was transferred into Gal-32 cells, the transformation frequency selecting in galactose was one twelfth to one fifth that of selecting in G418; with 10CR-1, the frequency was one fiftieth. A very likely explanation for the lower frequency of expression of the Gal+ gene compared to the SV2-neo gene is a difference in
promoter strength. Alternatively, the lower transformation frequency in galactose may be the result of DNA rearrangements in the presumably larger Gal+ gene. Furthermore, DNA methylation and changes in chromatin structure may decrease gene expression of transferred DNA [33].
It is concluded that we have isolated two human genomic cosmid clones (10CR-1 and 4CR-2) that correct the CHL Gal-32 mutation because 1) the genomic clones were constructed from primary transformant-3 (1-TR-3), which contains CHL and human DNA; for the reasons given above, the Gal+ phenotype of 1-TR-3 is due to the human DNA; 2) 10CR-1 and 4CR-2 grow in chloramphenicol and hybridize to the human Alu probe; thus, they contain human and camr DNA, derived from the pSV13-human library that generated primary transformant-3; 3) 10CR-1 and 4CR-2 transform Gal-32 to the Gal+ phenotype; whereas, six other camr, Alu-hybridizing, primary transformant clones did not; 4) 10CR-1 and 4CR-2 exhibit restriction fragments with the same size and with the same Alu-hybridizing properties, 5) mitochondrial protein synthesis is restored in Gal-32 cells transformed with clone 10CR-1 and could not be measured accurately with clone 4CR-2 due to instability.
Primary defect in Gal-32
The primary defect in Gal-32 causing the differential decrease in mitochondrial protein synthesis is unknown. It is unlikely that Gal- 32 is deficient in a general protein synthesis component such as mt ribosomal protein or mt aminoacyl synthetase because mutations in these components completely abolish all mt translation products [34,35]. A mutation altering tRNA structure, tRNA base modification, or tRNA charging might differentially affect the synthesis of mitochondrially encoded proteins according to their amino acid composition or nucleotide sequence, or unique context of a particular codon. In fact, the mouse mtDNA sequence reveals that there are more cysteine, aspartate, glutamate, and tyrosine residues in the subunits of NADH dehydrogenase and cytochrome c oxidase than in ATPase subunits[36]. Thus, a nuclear mutation affecting tRNACys, tRNAAsp, tRNAGlu or tRNATyr, might decrease cytochrome oxidase and NADH dehydrogenase peptides more than ATPase peptides, as is observed in Gal-32. Many disorders of the mitochondrion involve defects in the oxidative phosphorylation system, which comprises five multisubunit enzyme complexes encoded by both the nuclear and the mitochondrial genomes [37, 38]. Due to the multitude of proteins and intricacy of the processes required for a properly functioning oxidative phosphorylation system, identifying the genetic defect that underlies an oxidative phosphorylation deficiency is a formidable task [39]. Further investigation of the cloned human gene that corrects the Gal-32 mutation will yield information about its regulation, structure, and gene product and thereby enhancing our understanding of mitochondrial gene expression. This may also open possibilities for therapeutic modalities for those affected by mitochondrial disorders.
Human respiratory-deficient diseases
Deficiencies in human mt respiratory complexes have been associated with cardiac failure, stroke, skeletal muscle movement disorders, renal malfunction, diabetes, liver disease, Alzheimer’s disease, Parkinson’s disease, blindness, dementia, deafness, and myoclonic epilepsy (reviewed by Wallace [32]. Many of these diseases are maternally inherited due to missense, insertion-deletion, and copy number mutations in mtDNA. Mitochondrial tRNA mutations have been reported for myoclonic epilepsy and ragged-red fiber disease; mitochondrial encephalomyopathy, lactic acidosis, and stroke like symptoms; and maternally inherited myopathy and cardiomyopathy [32]. Like Gal 32, these tRNA mutations preferentially decrease cytochrome c oxidase and NADH dehydrogenase, which have the greatest number of mitochondrially encoded subunits.
The gene locus of Gal-32 is not known
Zheng et al. [40] have studied a lethal infantile mitochondrial disease due to deficiencies in the same respiratory complexes as Gal 32. NADH dehydrogenase and cytochrome c oxidase were drastically decreased in heart and skeletal muscle, but not brain; whereas, succinate-cytochrome c reductase activity was not greatly affected. Both parents were normal, suggesting a recessive, nuclear mutation, as in Gal 32. Gal-32 provides an intriguing animal model system for these human patients with analogous deficiencies in the mitochondrial respiratory complexes.
ZAS carried out most of the work in this study including the molecular biology and genetic studies: cloned the human Gal+ gene, conceived the theme of the project, designed and drafted the manuscript. CWB conceived the idea, participated in the design of the study and help draft the manuscript. All authors read and approved the final manuscript.
This research was supported by grants from the National Science Foundation; American Heart Association, Nation's Capital Affiliate; and Howard University Faculty Research Support Program. Another support includes Research Scientist Career Development Award from NIH to ZAS: # 5K01CA087554-04. We are grateful to Grace Mavodza for scanning the figures.