Drug Designing: Open Access
Open Access
ISSN: 2169-0138
ISSN: 2169-0138
Research - (2022)Volume 11, Issue 2
Objectives: To identify and select some of the existing molecules, with different therapeutic indications, as potential B-RAF inhibitors from literature. To explore and identify the appropriate B-RAF enzyme from the Protein Data Bank to carry out the molecular docking study on the selected molecules. To carry out the molecular docking studies on the selected molecules using molecular docking software AutoDock 4.2.2.
Methods: Molecular docking studies were conducted using AutoDock 4.2.2 version software. Antimicrobial agents [pyrazine-2-carboxylic acid derivatives (D1-D10)] and antifungal agents [albendazole derivatives (A1-A22)] were repurposed as BRAF inhibitors.
Results: Amongst the pyrazine-2-carboxylic derivatives compound D3 showed good results while amongst albendazole derivatives compound A12 showed promising results.
Conclusion: In silico drug designing was used to study pyrazene-2-carboxylic acid derivatives (D1-D10) and Benzimidazole (Albendazole) derivatives (A1-A22) as BRAF inhibitors. The online software named AutoDock version 4.2.2 was used to study molecular docking.
Cancer; Melanoma; Drug repurposing; Auto dock; BRAF gene; Antimicrobial; Antifungal
Drug discovery is a very long and expensive process. The main reason behind this is the high failure rate of drugs which can occur during any stage of drug discovery [1]. Over the years, many efforts have been made to ease this process and find cost-effective measures. One such development was molecular docking. Ever since its discovery in the 1980s, this technique has been increasingly used in the pharmaceutical industry [2]. It is a modeling technique which is used to study the interactions between a protein molecule and ligand; the protein usually is body enzymes, genes, etc., and ligand is the proposed drug molecule. Several software’s are available for molecular docking like Auto Dock [3], Auto Dock Vina [4], Schrodinger [5], GOLD [6] etc. They help understand the binding efficiency and conformation of the molecule. In silico drug design along with being cost effective, is also a very time-saving process, i.e., helps shorten the duration of drug discovery. In silico drug design is now a backbone of the pharmaceutical industry.
In the present research work, In silico drug design has been used to identify BRAF inhibitors which are an important class of anticancer agents. BRAF is a gene responsible for cell proliferation, apoptosis etc [7]. Its mutation is one reason for the development of various types of cancers in human beings like melanomas (50- 60%), colorectal cancer (10%), thyroid cancer (83%), non-small cell lung cancer (3%) and hairy cell leukemia (100%) [8-12,13]. Hence, BRAF inhibitors are considered as an effective class of anticancer agents. The existing drugs have various side effects like muscle pain, sunburn, joint pain, scaly skin, etc [14]. Moreover, their synthesis process is complex. Hence, the search for better inhibitors is essential.
Compounds with benzimidazole and pyrazine rings as nuclei in their structures are being frequently used in anticancer therapy [15- 23]. These rings have a striking similarity with the natural ligands, which makes them successful inhibitors. Several modifications have been studied on these rings over the years. Benzimidazole derivatives with pyrrolidine side chains are effective against hepatocellular cancer [24], 2-aryl derivatives against breast cancer [25], 1,2,5-trisubstituted derivatives against multiple myeloma, lymphoma and leukemia, etc., [26]. Similarly, pyrazine derivatives like pyrrolo [1,2a] pyrazine are effective against prostate and breast cancer [27], [1,2,4] triazolo[4,3-a] pyrazine derivatives bearing 4-oxo- pyridazinone moieties against cell lines A549, MCF-7 and HeLa and c-Met kinase [28], pyrazolo[3,4-b]pyrazines derivatives against breast cancer [29] etc,. Moreover, 2-(Allylthio)pyrazine derivatives [30], triazolo pyrazine derivatives [31], 3-Trifluoromethyl-5,6- dihydro-[1,2,4]triazolo Pyrazine Derivatives [32] etc. have been used to treat various types of cancers. Modifications of naturally occurring benzimidazoles and pyrazines with side chains like carboxamides and coumarin hybrids have also proved to be effective anticancer agents [33-35]. Apart from anticancer activity, certain pyrazine and benzimidazole ring derivatives have shown antifungal, antimicrobial, antitubercular, etc. activities as well [36- 41]. The main aim of this study is to identify such compounds and repurpose them as BRAF inhibitors.
BRAF gene and MAPK pathway
The BRAF gene is located on chromosome 7(7q34) and is responsible for encoding the BRAF protein [42]. It is a member of RAF kinase family. Its other isoforms are 1/c-RAF and A-RAF. All RAF proteins are substrates of MAPK (Mitogen activated protein kinase) Pathway [43]. These kinases have an important role in cell proliferation, inflammation regulation, apoptosis, differentiation, etc. [44] Alterations in RAF-MAPK pathways have been linked to various disorders like cancer, Noonan syndrome, LEOPARD syndrome, etc. gives a step-by-step overview of MAPK pathway [45] (Figure 1).

Figure 1: Overview MAPK Pathway.
Rationale for selecting the chemical class as BRAF inhibitors
Heterocyclic compounds are widely distributed in nature and are essential to life in various ways. Nitrogen-containing aromatic heterocyclic rings such as benzimidazole, pyrazine, and pyridine are important structural units in natural and synthetic pharmaceutical compounds. These heterocyclic rings have shown various pharmacological activities. Moreover, studies have shown heterocyclic compounds containing nitrogen, oxygen, and sulphur atoms are the core structures of several biologically active compounds. Amongst all heterocyclic compounds, nitrogen containing rings like benzimidazole, pyridine, piperidine, pyrazine, etc. are frequently employed citing their resemblance to nucleic acid bases [46,47]. This property is considered very useful when diseases with underlying gene mutations are to be treated. As mentioned above, gene mutations have been identified as one of the causes of cancer; just like BRAF mutation. In this case, a mutation in the V600E site of BRAF gene locks it in the active state. As a result, BRAF continuously keeps sending signals to MEK protein for cell proliferation. Phosphorylation of human protein kinases is an important mechanism for signal transduction, where adenosine triphosphate (ATP) acts as a phosphodonor. This indicates the importance of nitrogen-containing heterocyclic ring systems in disease therapy and explains the success of dabrafenib [48] and vemurafenib [49] as BRAF inhibitors as shown in Figure 1. Hence, it was decided to study the potential of some of the pyrazine and benzimidazole derivatives as B-RAF inhibitors (Figure 2).

Figure 2: Hypothetical model to predict the interaction of ligand (D3) with ATP binding site of enzyme B-Raf Kinase.
Softwares used
Chem sketch: It is a freeware, used for drawing and modifying organic and organometallic molecules. Molecules can be displayed in both two-and three-dimensional forms [50].
Open babel: It is software used to interconvert chemical file formats from .cdx to .pdbqt [51].
Auto dock version 4.2.2: AutoDock is an automated molecular docking software package that has been distributed to academics and non-profit research institutes free-of-charge since 1990. It is a suite of automated docking tools. It is designed to predict how small molecules, such as substrates or drug candidates, bind to a receptor of known 3D structure. AutoDock offers a variety of search algorithms to explore a given docking problem [52,53].
Python Molecular Viewer 1.5.6_17_14: Used for graphical visualization of Auto Dock Tools.
Selection of proteins
Among the several X-ray crystallographic structures of B-RAF kinase available on RCSB’s PDB, the enzyme structure having PDB ID: 4XV9 was selected for the study and downloaded from the Protein Data Bank (PDB). The X-ray crystallographic structure of B-RAF gene, 4XV9, was selected because it was found to be co crystallised with one of the highly active inhibitors PLX5568 and it has a resolution of 2.00 Å. The co-crystallised ligand, PLX5568 is a benzene sulphonamide derivative. It has been reported to inhibit B-RAF with an IC50 of 190 nM [54]. It shows the enzyme 4XV9 with its in-built ligands on the RCSB website (Figure 3).

Figure 3: 3-D view of 4XV9 with inbuilt ligand PLX 5568.
Ligand design
Literature survey and analysis of the unique ligand PLX5568 proved the anticancer activity of nitrogen-containing heterocyclic rings because of its resemblance to ATP molecule. It also indicated the importance of sulfonamide groups and electron withdrawing groups (like Cl, Br, F) in the molecules. Studies of the existing BRAF inhibitors, i.e.,dabrafenib and vemurafenib, also supplemented this data [55]. Accordingly, albendazole derivatives (A1-A22) and pyrazine 2-carboxylic acid derivatives (D1-D10) were selected as test ligands (Tables 1 and 2). Papers reported that these derivatives have been tested for antimicrobial and antifungal properties, respectively [56-58].
| Derivatives | Structure |
|---|---|
| A1 | 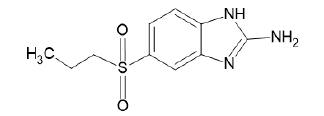 |
| A2 | 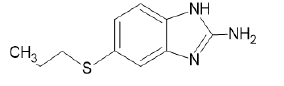 |
| A3 | 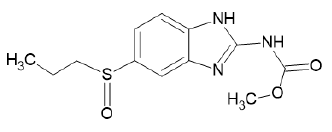 |
| A4 | 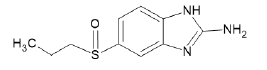 |
| A5 | 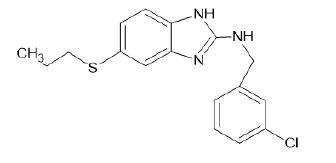 |
| A6 | 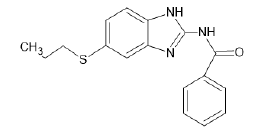 |
| A7 | 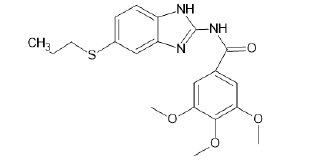 |
| A8 | 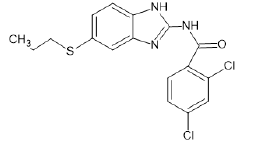 |
| A9 | 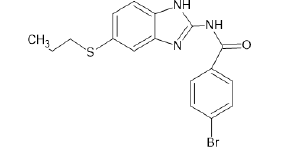 |
| A10 | 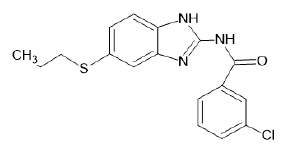 |
| A11 | 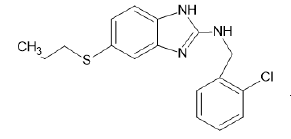 |
| A12 | 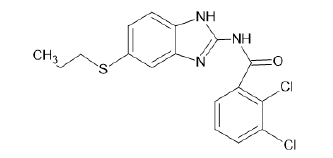 |
| A13 | 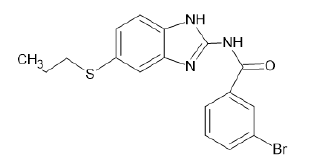 |
| A14 | 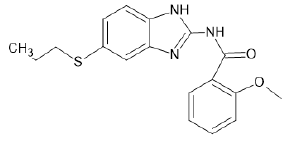 |
| A15 | 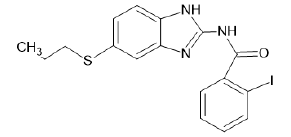 |
| A16 | 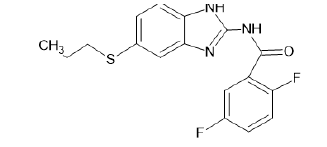 |
| A17 | 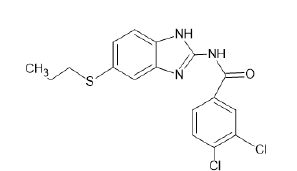 |
| A18 | 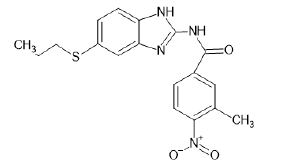 |
| A19 | 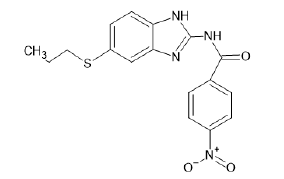 |
| A20 | 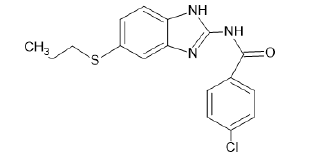 |
| A21 | 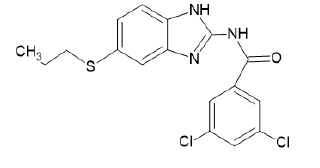 |
| A22 | 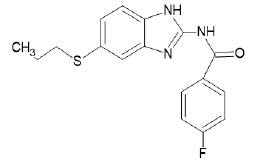 |
Table 1: Albendazole derivatives structures (A1- A22).
| Derivatives | Structure of derivative |
|---|---|
| D1 | 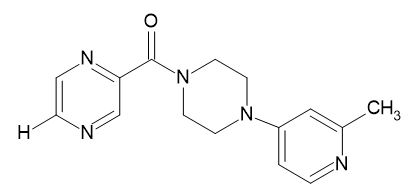 |
| D2 | 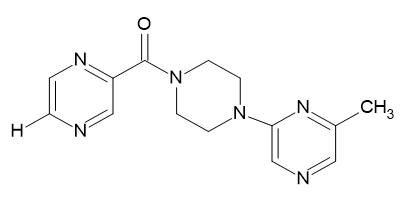 |
| D3 | 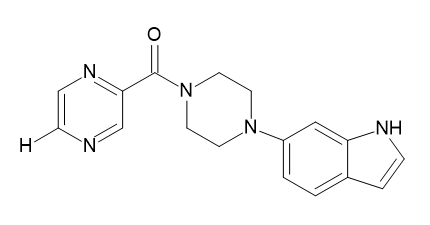 |
| D4 | 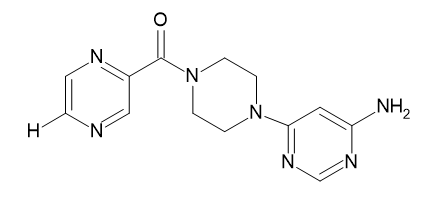 |
| D5 | 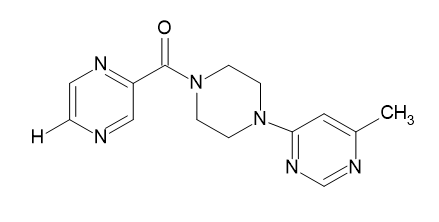 |
| D6 | 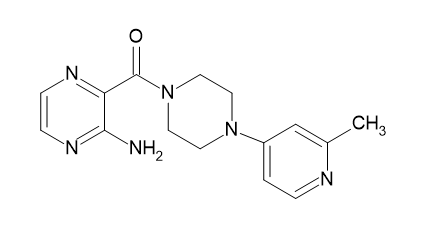 |
| D7 | 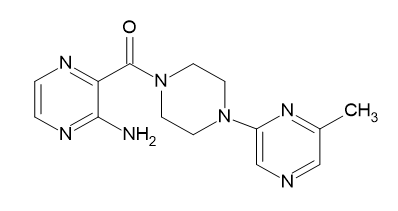 |
| D8 | 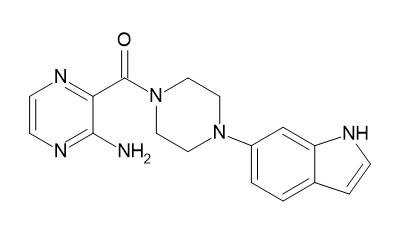 |
| D9 | 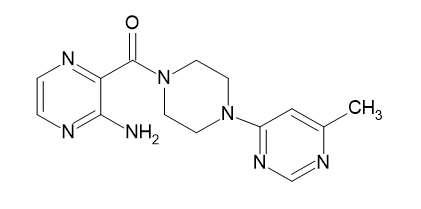 |
| D10 | 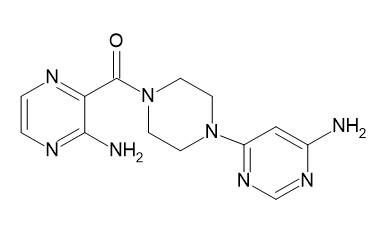 |
Table 2: Pyrazine 2- carboxylic Derivatives Structures (D1-D10).
Molecular docking studies on A1-A22 and D1-D10
Autodock 4.2.2 software was used as the software for the docking process. PMV Tool (Python Molecular Viewer) was as used as the interface.
Protein repairing and preparation: After dedocking the protein with its unique ligand and removal of water molecules, missing amino acid residues were searched for in the protein structure [59]. Through a literature search, it was understood that Lysine 522 was missing and the addition of this amino acid residue would improve the docking results [60]. Polar hydrogens were restored and Kollman charges were added and balanced. This prepared protein was saved in .pdbqt format. It shows the prepared protein molecule 4XV9 (Figure 4).

Figure 4: Screen shot of the repaired protein molecule 4XV9.
Preparation of ligand: The ligands were drawn using ChemSketch software and saved in .cdx (CS Chem Draw) format. These were converted to .pdbqt (protein data bank charge and torsion) format which is acceptable in AutoDock 4.2.2 software. After conversion, the ligands were loaded on the workspace of AutoDock. The required parameters like torsion, Kollman charge, gastegier charge, etc. were set up (Figure 5).

Figure 5: Preparation of ligand (Structure D3).
Grid selection: Process of blind docking was performed as the docking data of ligands (A1-A22 and D1-D10) for 4XV9 was not available. Grid size was selected as (100 Å) for this Autogrid module. The grid points giving the least binding energy were obtained. These coordinates were selected and for the final docking grid size 40 x 40 x 40 was used [61,62] it shows the grid selection process (Figure 6).

Figure 6: Generation of grid box.
Validation of the generated grid: After dedocking the in-built ligand (PLX5568) from the downloaded protein (4XV9); the ligand was redocked. The Root Mean Square Deviation value (RMSD) obtained was the same as that written on the RCSB website. This validates the docking protocol for further study [63].
Docking After successful grid generation, parameters were set with Genetic Algorithms, 40 conformations were tested for each ligand. The output was obtained as Lamarckain GA 4.2 format. The RMSD values of the structures were evaluated through a notepad. Depict the autogrid, autodock, and visualization of docking conformations after a successful docking process, respectively (Figures 7-9).

Figure 7: Screen shot of running of AutoGrid.

Figure 8: Screen shot of running of AutoDock.

Figure 9: Screen shot after playing the conformation.
After docking of the ligand and the enzyme (4XV9), the .dlg file is obtained, which contains complete information about the docked molecule. The derivatives of albendazole and pyrazine-2-carboxylic acid were docked to the enzyme 4XV9 using AutoDock tools 4.2.2 software (Tables 3 and 4).
It present the binding energy values of the docked molecules A1-A22 and D1-D10, respectively. The binding energies, no. of hydrogen bonds formed and RMSD values associated with the molecule were studied. The RMSD values were found to be 0. All compounds have formed hydrogen bonds in the range 1-2. The ligands which have not formed hydrogen bonds are A8, A9, A10, A13, A14, and A22. However, these ligands have shown good Van der Waals interactions.
It is observed from Table 3 that compound 1 with ligand ID A12 from the derivatives of albendazole, has a binding energy of 11.30 kCal/mole. It can be seen that this binding energy is more compared to the other compounds of the series. Thus, it can be concluded that the resulting interacting complex is very stable compared to the complex made by other compounds in the series. Binding energy is an important criterion for molecular docking analysis. It helps understand the specificity and sensitivity of the enzyme for the drug molecule. Lower binding energies are preferred. Most of the ligands showed hydrogen bonding with CYS 532. Some exceptions include compound A4 (H-bonding GLN 530, CYS 532), compound A6 (H-bonding with TRP 531), and compounds A15–A19 (H-bonding GLY 534, CYS 532). It shows ligand A7 docked at its binding site (Figure 10).
| Sr.No. | Ligand ID | Binding energy |
|---|---|---|
| 1 | A12 | -11.30 |
| 2 | A13 | -10.59 |
| 3 | A8 | -10.39 |
| 4 | A11 | -10.37 |
| 5 | A5 | -10.37 |
| 6 | A9 | -10.36 |
| 7 | A10 | -10.24 |
| 8 | A22 | -9.71 |
| 9 | A17 | -9.68 |
| 10 | A21 | -9.64 |
| 11 | A15 | -9.44 |
| 12 | A20 | -9.39 |
| 13 | A3 | -9.35 |
| 14 | A14 | -8.99 |
| 15 | A16 | -8.79 |
| 16 | A4 | -8.45 |
| 17 | A19 | -7.98 |
| 18 | A18 | -7.93 |
| 19 | A7 | -6.25 |
| 20 | A6 | -6.18 |
| 21 | A21 | -6.08 |
| 22 | A1 | -6.03 |
Table 3: Binding energy of the docked albendazole derivatives.

Figure 10: Docking of compound A7 (5-(propylsulfanyl)-1H- benzimidazol-2-amine) on 4XV9.
It is observed from Table 4 that Compound 1 with ligand ID D3 from the derivatives of Pyrazine-2-carboxylic acid has a binding energy of 11.21 kCal/mole. It can be seen that this binding energy is more compared to the other compounds of the series. Thus, it can be concluded that the resulting interacting complex is very stable compared to the complex made by other compounds in the series. All ligands have shown hydrogen bonding with CYS 532 except D6 which bonded with GLY534. It shows the ligand D3 docked at its binding site (Figure 11).
| Sr.No. | Ligand ID | Binding energy |
|---|---|---|
| 1 | D3 | -11.21 |
| 2 | D2 | -10.08 |
| 3 | D1 | -9.47 |
| 4 | D6 | -8.94 |
| 5 | D4 | -8.47 |
| 6 | D7 | -8.42 |
| 7 | D5 | -7.76 |
| 8 | D9 | -7.35 |
| 9 | D10 | -7.31 |
| 10 | D08 | -7.06 |
Table 4: Binding energy of the docked pyrazine 2-carboxylic acid derivatives.

Figure 11: Docking of compound A7 (5-(propylsulfanyl)-1H-benzimidazol- 2-amine) on 4XV9 Docking of compound D3 ([4-(6-aminopyrimidin-4- yl) piperazin-1-yl](5-methylpyrazin-2-yl)methanone) on 4XV9.
In the present research paper, an In silico drug design approach was used to study the potential of albendazole derivatives (A1-A22) and pyrazine-2-carboxylic acid derivatives (D1-D10) as BRAF inhibitors. The online software named AutoDock version 4.2.2 was used to study molecular docking. Amongst the pyrazine-2-carboxylic acid derivatives, D3 showed the best results with respect to binding energy 11.21 kCal/mol. While, amongst the albendazole derivatives, A12 showed the best results with binding energy 11.30 kCal/mol.
Thus, it can be concluded that D3 ([4-(1H-indol-6-yl) piperazin-1- yl) (5-methylpyrazin-2-yl) methanone) is the best B-RAF inhibitor amongst all tested compounds. Synthesis of this compound can be carried out using the procedure from the literature. Furthermore, this compound can be tested on mutant BRAF cell lines. In addition to this, enzyme inhibition studies can also be carried out to check their correlation with the results of In silico studies.
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar].
[Crossref] [Google Scholar].
[Crossref][Google Scholar] [PubMed].
[Crossref] [Google Scholar][PubMed].
[Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar].
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar]] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar].
[Crossref] [Google Scholar] [PubMed].
[CrossRef] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar].
[Crossref] [Google Scholar].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref][Google Scholar] [PubMed].
Citation: Kalia M, Parmar L, Pathak S, Patwa P, Shaikh Z, Ghadge O (2022) Repurposing Antimicrobial and Antifungal Agents as Potential BRAF Inhibitors. Drug Des. 11:204.
Received: 31-Jan-2022, Manuscript No. DDO-22-15703; Editor assigned: 04-Feb-2022, Pre QC No. DDO-22-15703 (PQ); Reviewed: 21-Feb-2022, QC No. DDO-22-15703; Revised: 28-Feb-2022, Manuscript No. DDO-22-15703(R); Published: 08-Mar-2022 , DOI: 10.35248/2169-0138.22.11.205
Copyright: © 2022 Kalia M et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.