Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Research Article - (2016) Volume 4, Issue 4
Platelets and von Willebrand factor (VWF) are recognized as important mediators of thrombosis at high shear rates, but their relative contributions are unclear. We employ a stenotic microfluidic test system to induce thrombus formation to occlusion on collagen at pathologic shear rates of 3500-6000 s-1. To obtain blood analogs with reduced platelet and VWF concentrations, human whole blood was diluted with saline by 90% or 99% and hematocrit restored by adding washed red blood cells. Platelets and VWF were selectively restored to normal levels. Blood from patients with known von Willebrand disease (VWD) was also investigated with and without the addition of VWF to investigate the contribution of platelet VWF. Normal whole blood led to channel occlusion in all tests (occlusion time, tocc=6.1 ± 2.2 min). 90% dilution with restored VWF occluded in 6/7 subjects even without restored platelets (tocc=16.6 ± 1.4 min). 90% dilution with restored platelets occluded in 2/5 subjects (tocc=27.2 ± 1.8 min), showing that added VWF is more effective for restoring occlusion than platelets. Addition of plasma VWF to VWD blood restored occlusion in only 1/4 severe subjects (VWF:RCo<15) and did not reduce normal occlusion time in the occluding subject, suggesting that VWF release from platelets plays an important role in high shear thrombosis. Logistic regression shows that VWF concentration and platelet count are strong predictors of thrombotic occlusion. Efforts to control high shear thrombosis may focus on VWF in addition to platelet function.
Keywords: Thrombosis; von Willebrand factor; von Willebrand disease; Platelets; High fluid shear rate
tocc: Occlusion time (s); VWD: von Willebrand Disease; VWF: von Willebrand Factor; VWF:RCo: Ristocetin cofactor assay
Myocardial infarction (MI) and ischemic stroke result from rapid, occlusive thrombus formation at the site of atherosclerotic plaque rupture [1]. Thrombus formation at atherosclerotic lesions occurs at high shear rates [2,3] and follows distinct pathways [4,5]. Briefly, collagen exposed by plaque cap rupture acts as a thrombogenic surface. At high shear rates, plasma von Willebrand factor (VWF) is adsorbed onto the surface. Platelets then bind to VWF at the A1 domain via platelet receptor GPIb. Bound platelets are activated by shear and/or soluble agonists and irreversibly bind via integrin αIIbβ3. Activated platelets release VWF at high local concentration, as well as other prothrombotic and proinflammatory factors, initiating a feed-forward amplification of thrombus growth. Clinical investigations have indicated that increased VWF levels are associated with increased risk of recurrent MI [6], MI in patients with angina pectoris [7], and stroke [8]. On the other hand, low levels of VWF are characteristic of von Willebrand Disease (VWD), a bleeding disorder with symptoms including nose bleeding, skin bruises and hematomas, prolonged bleeding from trivial wounds, oral cavity bleeding, and excessive menstrual bleeding [9].
It is well-accepted that platelets are a key constituent of arterial thrombi. Some investigators have shown that VWF is critical for platelet adhesion at high shear rates, but few studies have investigated the independent role of VWF in large-scale thrombus accumulation, particularly at high shear rates (>3000 s-1) relevant to MI and ischemic stroke. The objective of the present study is to determine the relative roles and minimum concentrations of plasma VWF and platelets in forming large-scale occlusive thrombus at high shear rates (>3000 s-1). Previously, we have theoretically shown that the number of exposed A1 domains on elongated, netlike VWF is likely to be the limiting factor in capturing circulating platelets [10]. We thus hypothesize that the contribution of VWF to high shear thrombus formation is equal to or greater than that of platelets. Further, we hypothesize that the high concentration of VWF released from platelets alpha-granules is important to the continued capture of platelets beyond the initial attachment to collagen. To achieve high shear rates with reasonable blood volumes, we employ a microfluidic device in conjunction with human-derived blood analogs containing controlled concentrations of plasma VWF, platelets, and red blood cells (RBC). We also study thrombus formation in blood from patients with known VWD to study the relative contributions of plasma VWF and platelet VWF. The results of this study lead to a better understanding of the roles of plasma VWF, platelet VWF, and platelet count in high shear thrombosis.
Blood collection and preparation
Blood was collected in either 60 mL syringes or 10 mL evacuated blood collection tubes (BD Vacutainer; Becton, Dickinson, and Company; Franklin Lakes, NJ) from normal volunteers and patients with known VWD under protocols approved by Georgia Institute of Technology and Emory University IRBs. Blood was anti-coagulated with 15.8 USP units/mL heparin. Blood analogs were produced by diluting whole blood from normal volunteers with normal saline at 90% and 99% dilutions.
Hematocrit was restored to 40% by adding washed red blood cells (RBCs). To test the contributions of VWF to high shear thrombosis, VWF (Humate-P; CLS Behring; King of Prussia, PA; gift from Hemophilia of Georgia) was added at 50, 100 and 200 IU/dL plasma, corresponding to the normal range of plasma VWF levels [11]. Humate-P has been shown to have a similar VWF multimer distribution as human plasma [12]. Isolated platelets were added to the blood analog at 120, 000 platelets/μL [13,14].
Additional experiments were conducted adding recombinant human ADAMTS-13 (R&D Systems, Inc.; Minneapolis, MN), which cleaves VWF [15], at normal concentration (0.75 mg/mL) [16] to 90% dilutions with restored VWF. One experiment was conducted with ADAMTS-13 added at six times normal concentration. All blood analogs were filtered under gravity at 50 μm using nylon mesh (McMaster-Carr Supply Company; Elmhurst, IL) immediately before experimentation. Table 1 summarizes the blood analogs considered. Additional experiments were performed with human fibrinogen (F4883; Sigma-Aldrich Co.; St. Louis, MO) added at normal concentration (300 mg/dL).
| Dilution | VWF Added | Platelets Added | |
|---|---|---|---|
| (IU/dL saline) | (103/µL) | ||
| Normal Whole Blood Control | 0% | 0 | 0 |
| Negative Control | 90% | 0 | 0 |
| Normal VWF and Normal Platelet | 90% | 100 | 120 |
| Normal Platelet Only | 90% | 0 | 120 |
| Normal VWF Only | 90% | 100 | 0 |
| Low Normal VWF Only | 90% | 50 | 0 |
| High Normal VWF Only | 90% | 200 | 0 |
| Normal VWF and Normal Platelet (99% dilution) | 99% | 100 | 120 |
| Normal Platelet Only (99% dilution) | 99% | 0 | 120 |
| Normal VWF Only(99% dilution) | 99% | 100 | 0 |
Table 1: Blood analogs produced by hemodilution.
Microfluidic test platform
To obtain high fluid shear rates with small blood volumes, blood was perfused through a stenotic microfluidic test section with a gradual contraction zone similar to an atheroma. Perfusion under a constant 65 mm pressure head yielded wall shear rates across 90% of the width of 3500-6000 s-1 in the stenotic region and 500-1000 s-1 in the non-stenotic region, corresponding to normal arterial wall shear rates [4]. The microfluidic channels were filled with 0.1% collagen Type I (fibrillar) solution (Chronopar; Chronolog, Inc.; Havertown, PA) and incubated at room temperature for 24 hours [17].
Data acquisition and analysis
The collagen-coated microfluidic chips were installed in the experimental set up shown in Figure 1. The discharge mass was measured to determine occlusion time, tocc, determined as the time from first blood contact with the stenosis to the time when the balance mass stopped increasing for at least 1 minute. Up to 5 mL of blood or blood analog was allowed to flow under a constant pressure head through the channel. Images of thrombus formation were acquired every 0.5 second using a microscope (DM6000 B; Leica Microsystems; Wetzlar, Germany) fitted with a high-resolution (1392×1020 pixels) CCD camera (Pixelfly; PCO; Kelheim, Germany). Incident light was filtered using a bandpass filter in the range of 480 nm (BG-42; Edmund Optics Inc.; Barrington, NJ). Image acquisition was facilitated by the μManager open-source microscopy software [18]. Blood from normal subjects collected in blood collection tubes was tested three times to characterize the experimental platform.
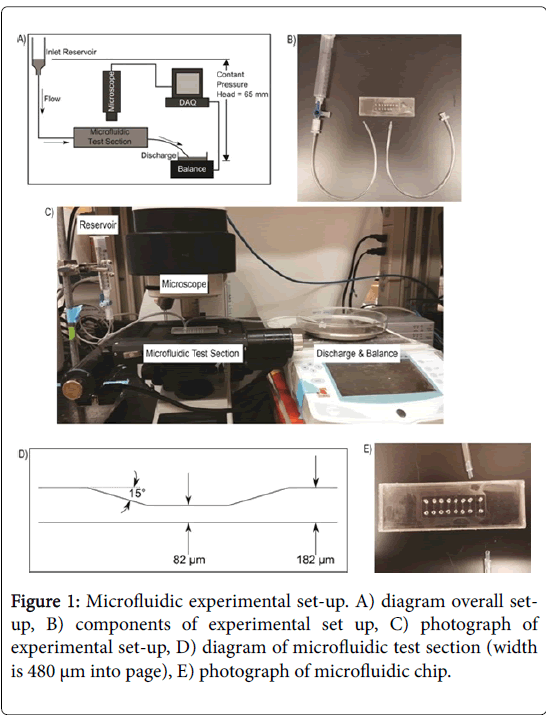
Figure 1: Microfluidic experimental set-up. A) diagram overall setup, B) components of experimental set up, C) photograph of experimental set-up, D) diagram of microfluidic test section (width is 480 µm into page), E) photograph of microfluidic chip.
Microscopy images were analyzed using MATLAB (R2013b; The Mathworks, Inc.; Natick, MA). First images were converted from 16-bit black-and-white to 8-bit color using the algorithm provided by the camera manufacturer [19]. For analysis, the total average intensity (grayscale) of the three color channels was used. A rectangular region of interest comprising the stenotic region was manually selected, and the total intensity (arbitrary units, AU) increase since the initial reference image (when blood first contacted the channel) was averaged over the area was computed for each time point. See Supplemental Data for more detailed methods.
Data is shown as mean ± standard deviation throughout. Statistical significance was evaluated at the 0.05 level, and data analyses were performed using SAS 9.3 (Cary, NC) and R Project (Vienna, Austria). Rates of occlusion and thrombus were calculated using frequencies and percents within control and successive blood dilation samples. Rate comparisons for occlusion and thrombus (yes vs . no) were made using an exact form of the Chi-square goodness of fit test. Time to occlusion was gauged across control and blood dilution groups using Kaplan- Meier curves and log-rank tests. Log-rank p-values were calculated overall and for each pairwise comparison using the Holm-Bonferroni method [20].
Samples that did not occlude were censored after 35 minutes of follow-up. Univariate and multivariable logistic regression was employed to identity significant risk factors associated with occlusion. Potential predictors included platelet counts and VWF at baseline and after dilution. Multivariable analysis was guided by the univariable results, and the final model was selected based upon the strongest predictors as indicated by statistical significance and model fit. Adjustments were made for within-sample, correlated data using Generalized Estimating Equations (GEEs). Predictive ability of independent variables was assessed via receiver operating characteristic curve (ROC) statistics, and model fits were evaluated with Quasi-Akaike Information Criterion (QIC) values [21].
Thrombus formation in normal subjects
Characteristic normal thrombus growth in the microfluidic test section is shown in Figure 2. Under grayscale imaging, the test section initially appears black as no thrombus has formed (Figure 2A). Thrombus begins to appear as patchy bright regions (Figure 2B and 2C). At occlusion, large thrombus deposits are clearly visible throughout the stenotic portion of the test section (Figure 2D). The change in total transmitted intensity across the test section is shown in Figure 2E.
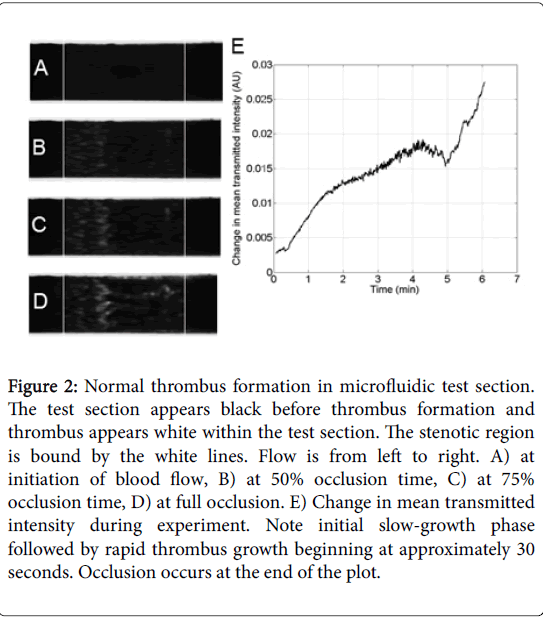
Figure 2: Normal thrombus formation in microfluidic test section. The test section appears black before thrombus formation and thrombus appears white within the test section. The stenotic region is bound by the white lines. Flow is from left to right. A) at initiation of blood flow, B) at 50% occlusion time, C) at 75% occlusion time, D) at full occlusion. E) Change in mean transmitted intensity during experiment. Note initial slow-growth phase followed by rapid thrombus growth beginning at approximately 30 seconds. Occlusion occurs at the end of the plot.
Initial slow deposition is followed by rapid thrombus accumulation. The final sharp increase in intensity may be due to clot contraction as the channel nears occlusion. The timescale of the intensity increase is in good agreement with previous results of the timescale of clot and platelet contraction of 8-12 min [22,23]. Characterization of occlusion time for normal thrombus formation is show in the Data Supplement.
All syringe-collected control cases led to channel occlusion measured by mass flow (n=12, p<0.001, Figure 3A) with a mean occlusion time of 6.1 ± 2.2 min. Normal subjects had platelets counts of 245 ± 66×103/μL (n=12) and VWF:RCo of 75 ± 69 IU/dL (n=6).
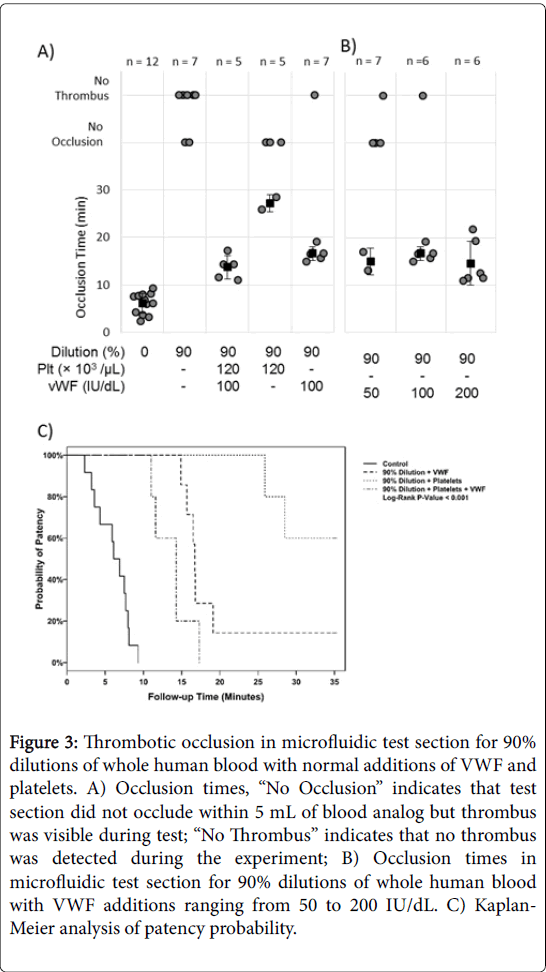
Figure 3: Thrombotic occlusion in microfluidic test section for 90% dilutions of whole human blood with normal additions of VWF and platelets. A) Occlusion times, “No Occlusion” indicates that test section did not occlude within 5 mL of blood analog but thrombus was visible during test; “No Thrombus” indicates that no thrombus was detected during the experiment; B) Occlusion times in microfluidic test section for 90% dilutions of whole human blood with VWF additions ranging from 50 to 200 IU/dL. C) Kaplan- Meier analysis of patency probability
Thrombus formation in 90% dilutions with selectively restored VWF and Platelets
None of the 90% dilution cases led to channel occlusion (n=7, p=0.016), indicating that dilution reduces the necessary components of high shear thrombosis to the point of eliminating thrombotic occlusion. 90% dilutions with restored platelets and VWF (100 IU/dL) occluded the channel in all cases (tocc=13.7 ± 2.5 min). Restoring only platelets to normal levels in 90% dilutions led to occlusion in 2/5 subjects (p=1) with the remainder showing visible, but non-occlusive, thrombus (tocc=27.2 ± 1.8 min). Restoring only VWF to 100 IU/dL in 90% dilutions restored occlusion in 6/7 subjects (p=0.125, exact test; tocc=16.6 ± 1.4 min, p<0.0001 compared to control, log-rank test). Thus, VWF is capable of forming occlusive thrombus, even with very low platelet counts. Restoring VWF alone also restored occlusion in more subjects and yielded a more rapid occlusion time than restoring platelets alone. The further addition of ADAMTS-13 to restored VWF analogs still resulted in high shear thrombosis that occluded in 4/5 subjects, and occlusion time was not reduced. Even adding ADAMTS-13 at six times normal concentration did not prevent occlusion (n=1). Thus, ADAMTS-13 does not appear to cancel the formation of VWF-mediated thrombosis in our high shear environment.
Kaplan-Meier analysis (Figure 3B) showed significantly lower probability of occlusion in the cases of 90% dilution compared to control (p<0.001, adjusted pairwise comparison). The addition of platelets to 90% dilution with normal VWF cases did not have a significant effect on occlusion probability (p=0.073, log-rank test). However, the addition of VWF to 90% dilution with normal platelet cases significantly increases the probability of occlusion (p=0.006, logrank test).
Kaplan-Meyer analysis suggests a trend towards lower probability of occlusion in conditions with decreased platelet count compared to conditions with decreased VWF concentration (p=0.054). Multivariable logistic regression for occlusion indicated that the resultant platelet count and resultant VWF concentration after dilution are significant predictors of occlusion (Table 2). The odds ratio (OR) for a 100,000 platelet/μL increase was 4.43, and the OR for a 100 IU/dL increase in VWF is 10.33. Together, platelet count and VWF concentration create a very good model for predicting occlusion (ROC=0.871). In 90% dilutions, added VWF was varied from 50 IU/dL to 200 IU/dL (Figure 3B).
| Characteristic | OR (95% CI) | P-Value |
|---|---|---|
| Platelet count (after dilution) | 4.43 (2.96 – 6.65)1 | <0.001 |
| VWF (after dilution) | 10.33 (2.51 – 42.50)2 | 0.001 |
| 1OR are based upon 100,000 unit increase in platelet characteristics 2OR are based upon 100 unit increases in VWF characteristics |
||
Table 2: Multivariable logistic regression for occlusion (yes vs. no).
At low-normal levels of VWF (50 IU/dL), 2/6 subjects occluded (p=0.688, exact test). When 100 IU/dL VWF was added to 90% dilution, 6/7 subjects occluded (p=0.125, exact test). At high-normal levels of VWF (200 IU/dL), 6/6 subjects occluded (p=0.031, exact test). Though additional VWF resulted in more subjects occluding at each subsequent experimental condition, the occlusion times remained rapid at the same mean times.
Thrombus formation in 99% dilutions with selectively restored VWF and platelets
Surprisingly, 99% dilutions with restored platelets and VWF (100 IU/dL) did not lead to thrombotic occlusion (Figure 4A). Rather, largescale thrombus formed, but did not fully occlude the vessel. Figure 4B shows the change in transmitted intensity for one such case, and rather than rapidly increasing as in Figure 2E, the curve shows a dynamic cyclic pattern with large peaks and valleys over a long experiment time. Note that the time scale is in tens of minutes to compress the time. The addition of fibrinogen to 99% dilutions with restored platelets and VWF resulted in full channel occlusion in 20.1 ± 1.6 min. Thus, fibrinogen did not enhance high shear thrombus growth rates, but did stabilize the large thrombus enough to cause flow occlusion.
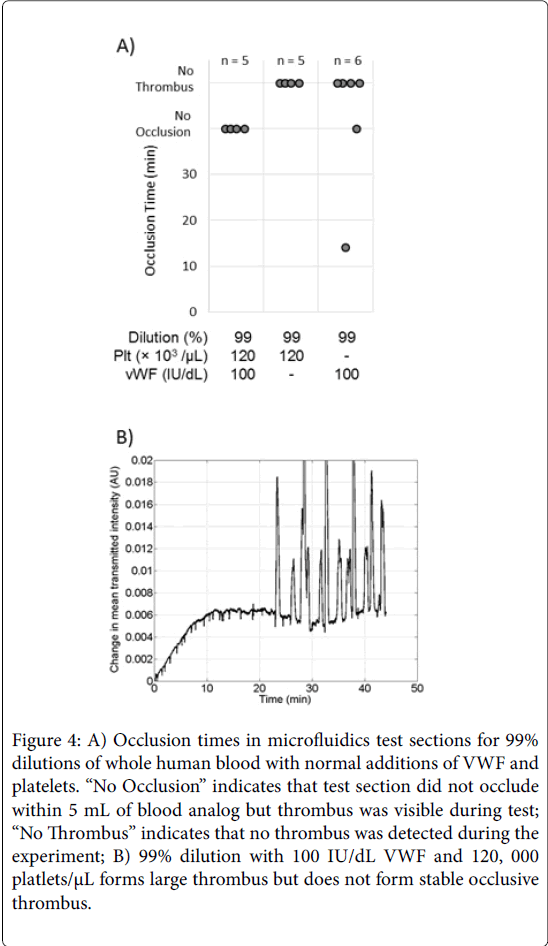
Figure 4: A) Occlusion times in microfluidics test sections for 99% dilutions of whole human blood with normal additions of VWF and platelets. “No Occlusion” indicates that test section did not occlude within 5 mL of blood analog but thrombus was visible during test; “No Thrombus” indicates that no thrombus was detected during the experiment; B) 99% dilution with 100 IU/dL VWF and 120, 000 platlets/μL forms large thrombus but does not form stable occlusive thrombus.
Thrombus formation in blood from VWD subjects
Studies were also performed on patients with VWD to isolate the effects of VWF released by platelets. Blood from VWD patients showed decreased occlusion compared to normal controls (Figure 5A). The normal blood for this comparison was collected in evacuated blood collection tubes for direct comparison with the VWD patient blood. All normal blood collected in evacuated blood collection tubes led to occlusion (n=15) with a mean occlusion time of 9.5 ± 3.4 min, slightly longer than the syringe blood controls, but not statistically different. Normal subjects had a mean platelet count of 275 ± 69×103/ μL. VWD patients had a mean platelet count of 280 ± 109×103/μL. VWD patients were separated post hoc into two groups for analysis. Mild VWD patients had values between 15 and 50 (15
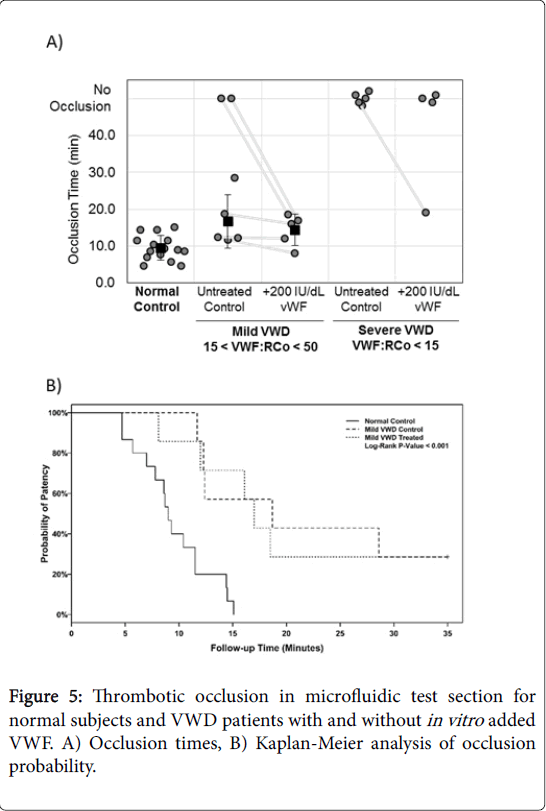
Figure 5: Thrombotic occlusion in microfluidic test section for normal subjects and VWD patients with and without in vitro added VWF. A) Occlusion times, B) Kaplan-Meier analysis of occlusion probability.
Thrombus formation under high fluid shear rate is mediated by platelets and VWF. In the present study, we utilized dilutions of whole blood to demonstrate that large-scale occlusive thrombus can form in microchannels with a normal concentration of VWF with very low platelet count. VWF was the primary mediator of high shear thrombosis, and VWF concentration and platelet count were strong predictors of occlusion. We also utilized blood from patients diagnosed with VWD to show that platelet VWF is important for occlusive thrombus formation.
Thrombus formation in 90% dilutions with selectively restored VWF and platelets
Human blood diluted to 90% with only normal concentrations of VWF added (100 IU/dL) led to thrombotic occlusion in most cases, despite having only 10% normal platelet count. Hence, VWF is capable of supporting high shear thrombosis even with few platelets. The occlusion times for 90% dilutions with added VWF were 2.7 times longer than for controls (16.6 ± 1.4 versus 6.1 ± 2.2 min). The increase in occlusion time may be due to the time required for sufficient platelets to be transported to the growing thrombus or to a decrease in VWF release by platelets. It should be noted, however, that the increase in occlusion time does not indicate that an equal number of platelets must be incorporated into the growth thrombus as in the whole blood case. If the same number of platelets were required to form occlusive thrombus, one would expect the occlusion time with 10% of normal platelet count to be 10 times as long as the whole blood control. The decreased platelet requirement may be facilitated by VWF selfassembly [25] and net formation [26], both processes that are enhanced by high shear rates. VWF nets have been proposed as a mechanism for rapid and efficient platelet capture at high shear rates [10] and may create thrombotic occlusion in blood analogs with very low platelet counts. 90% dilutions with only platelets restored occluded the microchannel in 2/5 cases with occlusion times >20 minutes, which closely recapitulates results for VWD samples. Logistic regression showed vWF concentration after dilution was more predictive of occlusion than platelet count after dilution.
Our results are consistent with Ogawa et al. [27], who showed that 40% hemodilution decreased thrombus onset and growth and that adding VWF accelerated thrombosis at shear rates of 1100 s-1. Note that hemodilution also reduces RBC count, which is known to reduce diffusive transport [28] and slow thrombus formation [29]. Additionally, the modest hemodilutions of 40% used in the Ogawa study retain high residual concentrations of plasma proteins and platelet count. The present study employs higher shear rates more relevant to arterial thrombosis, fixed hematocrit (40%) to control for changes in shear enhanced diffusivity, and larger dilutions to isolate and analyze the individual contributions of VWF and platelets.
Thrombus formation in 99% dilutions with selectively restored VWF and Platelets
When all other plasma proteins are diluted to 1% of their normal concentrations and VWF and platelets are restored, large thrombus formed, but did not occlude. These results suggested that an additional plasma protein(s) was necessary for thrombus stability and occlusion, but not growth. Addition of fibrinogen permitted stable occlusion of the channel. This finding is in agreement with Maxwell et al. [30] who noted that stable platelet aggregates do not form at shear rates of 1800 s-1 on a VWF-only matrix but do form on a VWF-fibrinogen matrix, indicating that fibrinogen plays a stabilizing role in thrombus formation at high shear rates.
Thrombus formation in blood from VWD subjects: The role of VWF release from platelets
Blood samples from subjects with known VWD were utilized to investigate the contributions of plasma and platelet VWF. VWD blood will have both low plasma VWF and low platelet VWF, whereas diluted samples have platelets with normal VWF content. While the in vitro addition of plasma VWF restored occlusion in both non-occluding samples with mild VWD, VWF addition did not restore occlusion in subjects with severe VWD. Thus, restoring plasma VWF alone is not sufficient to create occlusion if VWF is absent from the platelets. In contrast, restored plasma VWF and platelets with VWF do occlude. This indicates that platelet VWF is important for thrombotic occlusion at high shear rates.
The notion of VWF release for platelets acting as an important step in initiating rapid occlusive thrombosis is further supported by lag time and growth time data (See Data Supplement). A previous study by Turritto et al. showed that VWD blood perfused over rabbit subendothelium yielded normal platelet adhesion to the surface in some cases but reduced thrombus volume growth in the same cases [31]. These results support the conclusion of the present study that a moderate concentration of plasma VWF is sufficient for surfaceplatelet adhesion, but VWF release from platelets is required for largescale thrombus growth.
Our results support previous biophysics analysis that concluded that high VWF local concentration from release from platelets is needed for platelet capture at high shear rates [10]. Our results are also consistent with those of Moake, et al. [32], who used a cone-and-plate model to show that platelet aggregation under moderately high shear stress (up to 60 dynes/cm2 or a shear rate of approximately 1700 s-1) is mediated by either plasma VWF or VWF released from platelets. Isolated platelets re-suspended in buffer showed little aggregation, but VWF added to the suspension induced aggregation under shear while severe VWD platelets in buffer showed no aggregation. The present study extends these results to a single-pass system at higher shear rates that better mimic the shear conditions of a stenotic artery. Whereas the Moake et al. study investigated only small platelet aggregates, the present study used large occlusive thrombus formation as the endpoint to better model clinically relevant thrombi. The major finding of the present study is that normal VWF can lead to thrombotic occlusion even with small platelet counts, showing the dominance of VWF in high shear thrombosis. This is even more surprising as many would guess that platelet bulk would be critical to creating the mass of the thrombus. Taken together, it appears that a threshold of plasma VWF is needed to initiate thrombosis at high shear rate, while VWF release from platelets produces large, rapid occlusive thrombosis.
A previous study in mice showed that mice deficient in platelet VWF could support hemostasis based on tail bleeding time, and platelet VWF could reduce bleeding time in some VWF-deficient mice [33]. Microfluidic experiments in the same study showed that either plasma or platelet-derived VWF supported platelet adhesion to a collagen surface at shear rates of 2000 s-1. However, hemostasis was the focus of the previous study and large-scale platelet thrombus was not studied. The results of the present study show that while plasma VWF alone may support surface adhesion of platelets, VWF release from platelets may be required to create large-scale occlusive thrombus at high shear rates. Platelet-derived VWF has also been shown to have a higher molecular weight distribution than plasma VWF, and high molecular weight VWF is more hemostatically active [34]. The same study showed that VWF released by platelets is resistant to cleavage by ADAMTS-13, which may further contribute to rapid thrombus growth leading to occlusion. Our results concur that ADAMTS13 does not prevent occlusive thrombus formation with either plasma or platelet derived VWF.
Clinical implications
The results of the present study have clinical implications for the prevention of arterial thrombosis. Presently, anti-platelet agents, primarily aspirin and P2Y12 inhibitors (e.g. clopidogrel, prasugrel, and ticagrelor) are the treatments-of-choice for long-term therapy for their relatively low cost and oral administration. That thrombus may form with only 10% of normal platelet count may explain some of the documented aspirin and clopidogrel non-response [35,36]. The effectiveness of anti-platelet agents may be in part the result of decreased platelet activation leading to decreased VWF release from platelets. Non-responsiveness may also be due to platelet activation via alternative pathways, such as shear [37,38] or GPIb binding [39]. Alternative anti-thrombotic agents that target VWF are currently under development [40] and may provide an additional or alternative treatment for patients who are non-responsive to anti-platelet drugs. The present results may also have important implications for bleeding in surgery or trauma. In case of severe bleeding with saline fluid replacement, the blood undergoes hemodilution, which could lead to reduced hemostasis. Our results indicate that replacing only platelets may be insufficient for restoring hemostatic function as plasma VWF may be of greater importance at high shear rates..
Limitations
The present study has several limitations. Blood was anticoagulated with heparin for transport, which reduces the contribution of fibrin formation in thrombus formation. Sodium citrate with subsequent recalcification instead of heparin could be used to retain fibrin formation. For some studies, blood was collected in blood collection tubes, which induce higher shear rates and may prematurely activate platelets. This was controlled for in the present study, but may lead to differences in thrombus growth and occlusion times compared to in vivo thrombosis. Multiple platelet counts were not investigated. Calcium ion concentration was not controlled or restored in hemodilutions, which may impair platelet activation. The VWF concentrate also contained coagulation Factor VIII. The addition of Factor VIII is not expected to have a large effect on the results since the blood was anticoagulated with heparin.
In conclusion, VWF can lead to flow occlusion by high shear thrombosis, even with significant dilution of platelets and other plasma proteins. Plasma VWF can support occlusive thrombus formation even with low platelet counts, but is insufficient if platelet VWF is absent in the case of severe VWD. For flow occlusion from a stable thrombus, approximately 10% fibrinogen level appears to be needed. Greater understanding of the role of plasma and platelet-derived VWF may lead to more effective prevention of high shear thrombosis.
We gratefully acknowledge J. Rhett Mayor (GWW School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA) for the micro-machining of molds for the fabrication of microfluidic test sections. This work was supported by a grant from the Center for Pediatric Innovation, Georgia Institute of Technology, Children’s Healthcare of Atlanta, and Emory University; Lawrence P. Huang Chair Funds, Georgia Institute of Technology; and the John and Mary Brock Discover Fund. L.D.C.C. was supported by an American Heart Association Pre-Doctoral Fellowship (14PRE18080005).