Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Research Article - (2014) Volume 2, Issue 1
Background: Consanguinity remains common in several populations around the world, and varies from country to country. In Pakistan, close consanguineous unions continue to be extremely common as in South West Asia. Here, we describe the frequency of Rare Bleeding Disorders (RBDs), their types and clinical features among patients seeking advice for bleeding tendencies from a single centre in Pakistan. Patients and Methods: Pre-designed data sheets were filled by incorporating patient’s demographics, family history, present and past history of bleeding episodes with the associated signs and symptoms. In female cases, maternal and obstetrical history was taken. Blood samples were collected for Complete Blood Count (CBC) and coagulation assays. Results: Out of 600 patients diagnosed with inherited coagulation bleeding disorders, 64 subjects had RBDs (11%). Among them, 35(55%) were male and 29(45.31%) were female. Median age of patients was 9.8 years, (range, 12 days to 37 years). History of consanguinity was present in 85% of cases and significant family history of bleeding in 54% of patients. The most common deficiency was FXIII (n=18, 28%) and FVII deficiency (n=18, 28%) followed by fibrinogen deficiency (n=15, 23%), FV deficiency (n=5, 8%), FX deficiency (n=4, 6%), FXI deficiency (n=2, 3%) respectively. There were one case of each combined FV and VIII deficiency and vitamin K dependent factor deficiency (2%). Clinical bleeding episodes were classified into four categories according to severity: grade III and II bleeding were noted in 53%, and 47% patients, respectively. Menorrhagia (81%), gum bleeding (46.8%), easy bruising (42.1%), umbilical cord bleeding (35.9%), epistaxis (32.8%), hematomas (31.2%), hemarthrosis (26.6%) and delayed wound healing (25%) were the main clinical manifestations. Fresh frozen plasma/cryoprecipitate was used in the management of most patients. Conclusion: The study shows that autosomal recessive disorders are common in the setting of consanguineous marriages. Further studies of the association between phenotype and genotype in this subset of patients are needed.
Keywords: Rare bleeding disorders; Autosomal recessive disorders; Consanguineous marriages; Pakistan
The Rare Bleeding Disorders (RBDs) are inherited deficiencies of coagulation factors such as fibrinogen, prothrombin (FII), FV, FVII, FX, FXI, FXIII and of more than one factor such as combined FV and FVIII deficiency and deficiency of vitamin K dependent factors. They are usually transmitted in an autosomal recessive manner and represent 3% to 5% of all inherited coagulation deficiencies [1,2].
Autosomal recessive traits and severe deficiencies are more likely to be found in populations where marriage between blood relations (consanguineous marriages) is common and in rare cases individuals may inherit more than one disorder. The incidence of this deficiency is higher in countries with high rates of consanguineous marriages [3]. In Pakistan, close consanguineous unions continue to be extremely common as in South West Asia. As a result of these marriages, an increased rate of congenital anomalies and autosomal recessive disorders become common and run in close families and tribes [4]. They are so frequent in these areas, that they even exceed the prevalence of disorders like hemophilia B in those areas [5].
Bleeding pattern may considerably vary between affected individuals of RBDs i.e. from deficiency to deficiency and from patient to patient. Patients usually present at early neonatal period with prolonged umbilical stump bleeding, post circumcision bleeding, bruises, mucosal hemorrhage, cerebral bleeding, hemarthrosis, hematoma, intracranial hemorrhage, epistaxis, menorrhagia and joint bleeding. However, life-threatening bleeding such as central nervous and musculoskeletal bleeding appears to be less frequent than in hemophilia [6].
Here, we describe the incidence of rare inherited coagulation bleeding disorders, their types and clinical features among patients seeking advice for bleeding tendencies from a single reference centre in Pakistan and to compare these data to those reported in other populations.
The study was approved by an institutional ethics committee and was done in accordance with the declaration of Helsinki. Informed written consent was obtained from all adult subjects, parents or legal guardians in local language. This was a cross-sectional, descriptive, epidemiological study carried out at the National Institute of Blood Disease (NIBD) and bone marrow transplantation (a reference tertiary care centre for patients with bleeding and hematological disorders across the country) from August 2010 to February 2013. Patients with a history of congenital bleeding disorders or a suspected bleeding tendency were screened and only RBDs patients were enrolled.
Pre-designed data sheets were filled by incorporating patient’s demographics, family history, present and past history of bleeding episodes with the associated signs and symptoms. In female cases, maternal and obstetrical history was taken. The RBD database www.rbdd.org was used for this purpose and bleeding graded according to Peyvandi et al. [7].
Blood samples were collected for Complete Blood Count (CBC), DNA extraction in EDTA and 0.109 M (3.2%) trisodium citrate in a ratio of 9:1 for coagulation assays. Prothrombin Time (PT), Activated Partial Thromboplastin Time (APTT), fibrinogen and factor assays were performed. Urea clot solubility test in 5 M urea and FXIII assay was done in cases where all coagulation tests were normal. Samples were saved for antigen levels & DNA for mutational analysis.
The normal values of PT and APTT were 11-15 seconds and 25- 32 seconds, respectively. Different coagulation factors assays were performed on the basis of initial testing. Factor XI assays were APTT based while factors V, VII and X assays were PT based by one-stage assay. Diagnostic Stago (France) deficient plasma and other reagents were used for factor assays. Reference values for all APTT based factor activity assays were taken between 60% and 150% and those for PT based factor activity assays were taken between 70% and 150% that have been set by the kit provider. For FXIII activity reference range was 69-143% of normal.
The statistical package SPSS-17 was used to analyze the data. Frequency and percentage were computed for categorical variables and mean and Standard Deviation (SD) was estimated for quantitative variables. Similarly mean and SD was reported where data was distributed normally otherwise median and ranges were reported. The chi-square test was applied to calculate significance.
Out of 600 patients diagnosed with inherited coagulation bleeding disorders, 64 subjects had rare bleeding disorders (11%). Among them, 35 were male and 29 were female. Median age of patients was 9.8 years, (range 12 days to 37years) whereas the median age of onset of the first episode of bleeding was 6 months (range, from birth to 25 years). History of consanguinity was present in 85% of cases and significant family history of bleeding in 54% of patients.
The most common deficiency was FXIII (n=18, 28%), FVII (n=18, 28%), followed by fibrinogen (n=15, 23%), FV (n=5, 8%), FX (n=4, 6%) and FXI deficiency (n=2, 3%), respectively. There was one case (2%) of combined FV and VIII deficiency and 1 case of combined vitamin K dependent factor deficiency (Figure 1). There were 14 patients with severe FXIII deficiency (activity 3.8% ± 1.8 ) and 4 related family members with a bleeding history (activity 43.5% ± 18.6). In patients with FVII deficiency, 13 had levels <5%, 3 patients had levels >5-10% and 2 patients had >10-30%. Thus its severity was graded as per the clinical presentation [8,9]. All patients with fibrinogen deficiency were diagnosed with afibrinogenaemia (less than 20mg/dl). The patient with FV and FX deficiency had levels <5%. Patients with other RBDs had a mild phenotype.
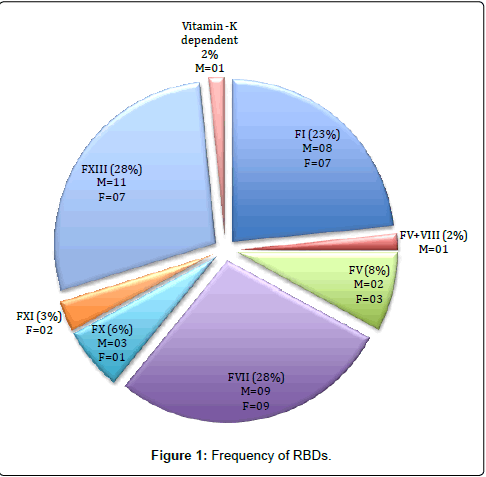
Figure 1: Frequency of RBDs.
Clinical bleeding episodes in RBDs were classified into four categories according to severity: grade III and II bleeding were noted in 53% and 47% patients, respectively. Menorrhagia (81%), gum bleeding (46.8%), easy bruising (42.1%), umbilical cord bleeding (35.9%), epistaxis (32.8%), hematomas (31.2%), hemarthrosis (26.6%) and delayed wound healing (25%) were the main clinical manifestations.
A total of 6 patients (9.3%) developed intracranial hemorrhage (ICH) (Table 1). Factor XIII was the most common factor deficient in patients with ICH, found in 3 (16.6%) patients followed by two patients with FX and one with FVII deficiency.
Patients with FXIII deficiency presented with umbilical cord bleeding (60%), menorrhagia (60%), hematoma (44.4%), bruises (33.3%), delayed wound healing (22.2%), gum bleeding (22.2%), epistaxis (22.2%) and ICH (16.6%). Two female patients besides having menorrhagia also had a history of miscarriage. In this study we have only seen patients with severe fibrinogen deficiency (<20 mg/dl), the most frequent bleeding symptoms were gum bleeding (60%), umbilical cord bleeding (53.3%), menorrhagia (50%), bruises (40%), epistaxis (33.3%), hemarthrosis (26.6%) and hematoma (26.6%) (Table 1).
| Factor Deficiency | FI | FV | FVII | FX | FXI | FXIII | FV+ FVIII | Vitamin K dependent | ||
| Bleeding severity | GradeII | GradeII | GradeIII | GradeIII | GradeII | GradeII | GradeII | GradeII | Total n(%) | ºBleeding Grades |
| Symptoms | ||||||||||
| Gum Bleeding | 9(60%) | 5(100%) | 7(38.8%) | 2(50%) | 1(50%) | 4(22.2%) | 1(100%) | 1(100%) | 30(46.8) | II |
| Bruises | 6(40%) | 5(100%) | 5(27.7%) | 2(50%) | 2(100%) | 6(33.3%) | - | 1(100%) | 27(42.1) | II |
| Epistaxis | 5(33.3%) | 4(80%) | 8(44.4%) | - | - | 4(22.2%) | - | - | 21(32.8) | II |
| ICH | - | - | 1(5.56%) | 2(50%) | - | 3(16.6%) | - | - | 06(9.3) | III |
| Menorrhagia | 1(50%)out of 2* | 2(100%)out of 2* | 7(100%)out of 7* | - | - | 3(60%)out of 5* | - | - | 13(81)out of 16* | II |
| Repeated Abortions | 1(50%)out of 2* | - | - | - | - | 2(40%)out of 5* | - | - | 03(18.7) out of 16* | II |
| Umblical cord bleeding | 8(53.3%) | 2(40%) | 1(5.56%) | 2(50%) | - | 10(60%) | - | - | 23(35.9) | III |
| Wound Healing | 6(40%) | 1(20%) | 4(22.2%) | - | - | 4(22.2%) | - | 1(100%) | 16(25) | II |
| Hematuria | - | 1(20%) | 2(11.1%) | - | - | - | - | 03(4.69) | III | |
| Circumcision | 2(25%)out of 8** | 1(50%)out of 2** | 0 out of 9** | 0 out of 3** | - | 2(18.18%) out of 11** | 0 out of 10** | 0 out of 10** | 05(14.2) out of 35** | II |
| Hematoma | 4(26.6%) | 1(20%) | 5(27.7%) | 2(50%) | - | 8(44.4%) | - | - | 20(31.2) | III |
| Hemarthrosis | 4(26.6%) | 2(40%) | 6(33.3%) | 2(50%) | - | 3(16.6%) | - | - | 17(26.6) | III |
| GI Bleeding | 1(6.66%) | 1(20%) | 1(5.56%) | - | - | - | - | - | 03(4.69) | III |
| Hemoptysis | - | - | 2(11.1%) | - | - | - | - | - | 02(3.13) | III |
| Melena | 1(6.66%) | - | 4(22.2%) | - | - | - | - | - | 05(7.81) | III |
Table 1: Phenotype Characteristics of RBDs patients.
We analyzed data to identify the correlation of symptom severity with consanguineous marriages. A consanguineous marriage was defined as a marriage between people who were second cousins or closer. Patients who had history of consanguinity were more in number and were affected by grade II and III bleeding than those who had no history of consanguinity. Chi-square test was applied and results were found significant in all RBDs with positive consanguineous marriages (Table 2).
| POSITIVE CONSANGUINEOUS MARRIAGES | ||
| Factor | Grading | Total |
| I | Grade II | 15 |
| V+VIII | Grade II | 1 |
| V | Grade II | 3 |
| VII | Grade III | 14 |
| X | Grade III | 4 |
| XI | Grade II | 1 |
| XIII | Grade II | 17 |
| Total | 55 | |
| NEGATIVE CONSANGUINEOUS MARRIAGES | ||
| VII | Grade III | 1 |
| FXIII | Grade II | 1 |
| Total | 02 | |
Table 2: Correlation between symptom severity and consanguineous marriages.
Fresh frozen plasma / cryoprecipitate (10-15 ml/kg) and antifibrinolytics (10 mg/kg/dose 3-4times/day) were used in the management of most patients on demand. A total of 392 bleeding episodes were identified during different time periods requiring visit to hospital, among them 243 were daycare, 35 ward admissions while 114 were OPD visits. FVII deficiency was the most common factor deficiency with hospital visits for gum bleeding; epistaxis, hemarthrosis, menorrhagia and ICH were the main bleeding manifestations requiring hospital visit and admission. FXIII deficiency was the second common with umbilical cord bleeding, epistaxis, menorrhagia, hematomas, hemarthrosis, gum bleeding, circumcision and ICH needing hospital visit and admission followed by FX deficiency with, umbilical cord bleeding, hemarthrosis gum bleeding, hematoma and ICH. Then was fibrinogen deficiency with day care and outpatient visits for gum bleeding, circumcision, hematoma, delayed wound healing, epistaxis and hemarthrosis.
Patients with congenital clotting factor deficiencies have a lifelong risk of bleeding ranging from minor to cataclysmic hemorrhages. This is the first large study from Pakistan investigating RBDs, with emphasis on the clinical manifestations and pattern of bleeding. There is lack of information on the prevalence and features of these disorders from this area except a few local studies showing the frequency and clinical spectrum of coagulation factors [10,11]. A high incidence of consanguinity in Pakistan [12], also observed in the study, leads to an increased incidence of autosomal recessive disorders like RBDs 10.6%, which is almost double the number, reported for all inherited coagulation deficiencies (3-5%).
NIBD receives patients from different geographical regions of Pakistan from different ethnic backgrounds. So in essence the group of patients we examined in this study came from a mix of ethnicity including Sindhi, Punjabi, Balochi and Urdu speakers. However, this study did not capture the data on ethnicity particularly, so that remains a limitation of this analysis.
Comparison of data obtained from this study with data from the World Federation of Hemophilia (WFH) and International Rare Bleeding Disorders Database (RBDD) shows that FVII and FXI deficiencies are the most prevalent (29-33%) among RBD patients followed by fibrinogen, FV and FX deficiencies (8-9%), FXIII around (6%) and combined FV and FVIII 5% [6,7]. Some local studies of RBDs showed that FVII, FXIII and FX deficiencies are similar to what has been observed in our neighboring countries like India and Iran [13,14]. However in India, there is regional variation. In Northern India FX is common whereas in Southern India FXIII is most common [15].
In the study patients with history of consanguinity were more symptomatic than those with no history of family marriage. The probability of compound heterozygosity is low in consanguineous patients and the probability of having a “mild” mutation out of 2 (compound heterozygotes) is higher than out of 1 (homozygotes). In a patient who is compound heterozygous, one “mild” mutation along with a “severe” mutation would have a mild phenotype or would be asymptomatic. Therefore, patients born from consanguineous marriages could be statistically more “symptomatic” than the others.
Despite their high prevalence in our population, these disorders often remain undiagnosed, and particularly so unless significant bleeding occurs. Muco-cutaneous bleeding (such as gum bleeding, easy bruising, epistaxis and menorrhagia) was the most common presenting features seen in the majority of patients (70%). These symptoms were divided into three grades according to their severity [7]. In our study, FXIII, FVII and FX were the most severe bleeding disorders. In FX deficient patients, grade III bleeding occurred in half of the patients with ICH in 2 patients with severe FX deficiency. Later one of these patients developed hemarthrosis of the knee joint. Another 2 patients had recurrent gum bleeding, hematoma, umbilical cord bleeding, hemarthrosis and easy bruising (Table 1). These clinical findings are comparable to those observed in FX deficient Indian, European and South American patients [16] where muco-cutaneous bleeds were 82% with ecchymosis occurring more frequently than epistaxis [13]. However we had less FX deficient patients (6%) in our study. In the Iranian study epistaxis was most common 72% followed by hemarthrosis in 69%, umbilical stump bleeding in 28 % and CNS bleed in 9% [17].
FVII deficiency was the most common and severe deficiency in our study with grade III bleeding (53.3%) requiring a hospital visit in our study. Menorrhagia and epistaxis, were the most common symptoms followed by gum bleeding hemarthrosis of knee joint, hematomas, malena, hemoptysis, hematuria and ICH were also significant in our patients. These findings are similar to what has been observed in Iranian and Italian patients [14]. In severe FVII deficiency, ICH is common and has been reported from 15% to 60 % of cases [18], though only one patient presented with cerebral bleed (6%) in this study. Our data of FVII deficient patients also shows that factor VII levels correlate poorly with bleeding risk. Some patients with undetectable FVII are asymptomatic while others with detectable levels had grade III bleeding as previously stated in the literature [19].
Patients with FXIII deficiency presented with umbilical cord bleeding, hematomas, menorrhagia, bruises, delayed wound healing, gum bleeding, epistaxis and ICH. Two female patients besides having menorrhagia also had a history of miscarriage (Table 1) one of these patients have a homozygous FXIII mutation while the other is a heterozygous. In Indian patients with FXIII deficiency, ecchymosis was the most common symptom [20]. The bleeding manifestation observed in our patients are more similar to those observed in the Iranian population [21]. Similarly the incidence of ICH has been reported to be around 25-30%. The incidence in our series was lower because of lack of diagnosis and due to the limited availability of the urea clot solubility and FXIII test in different centers. In this study FXIII deficient patients had less hospital visits as compared to FVII patients because eleven FXIII patients were referred to us for diagnosis and management from remote areas of Sindh.
Fibrinogen abnormalities include afibrinogenaemia, hypofibrinogenaemia and dysfibrinogenaemia. Dysfibrinogenaemic and hypofibrinogenaemic patients are usually asymptomatic, while those with afibrinogenaemia have a bleeding tendency that usually manifests in the neonatal period [3,6]. In this study we have only seen patients with severe fibrinogen deficiency (<20 mg/dl) with the most frequent bleeding symptoms being gum bleeding, umbilical cord bleeding, menorrhagia, bruises, epistaxis, hemarthrosis and hematoma. We were unable to perform antigen levels to distinguish dysfibrinogenemia from others. In fibrinogen deficiency patients, overall, grade II type of bleeding was observed 50% with fewer hospital visits and admission as compared to other RBDs in the study. This is in contrast to the EN-RBDD results which showed patients with FXIII, fibrinogen, and FX had the highest proportions of patients with grade III bleeding [7]. That is the reason why we need local RBD registry to compare our data with other populations.
In our series, we had 5 patients with FV deficiency having mucocutanous type mainly gum bleeding, epistaxis, easy bruising and menorrhagia. There were few patients with other RBDs like FXI deficiency was found in 2 patients with grade II bleeding (gum bleeding and easy bruising). One patient had combined FV+FVIII deficiency and one patient had vitamin K-dependent factor deficiency of FII, FVII, FIX, FX mainly with muco-cutanous type of bleeding and milder symptoms. These findings correlated with the findings from Iranian and Indian cohorts [13,22,23].
The study shows that autosomal recessive disorders are common in a setting of consanguineous marriages. This keeps all the beneficial and adversely affecting recessive genes within the family; in a homozygous state, which results in life threatening diseases. FXIII, FVII and fibrinogen deficiencies are the most common among the RBDs in our population. FX, FVII and FXIII were the most severe disorders with the highest proportion of grade III & grade II bleeding episodes. Mucocutaneous type of bleeding was the most common presenting feature seen in the majority of patients.
Thus further studies of the association between phenotype and genotype in this subset of patients are needed to prevent the spread of such common occurrences of these rare disorders in the community. Knowing the incidence of these rare bleeding disorders will aid the diagnosis and management of facilities throughout the country.