Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2008) Volume 1, Issue 2
The proper theoretical description of the distribution of the node degree for yeast protein-protein interaction network was investigated to deal with the observed discrepancy between usually proposed models and the existing data. The power law or the generalized power law with exponential cut-off were shown to be inaccurate within a wide range of degree values. Proposed linear-combination-of-exponentialdecays- method exactly characterizing the distribution by the spectrum of decay constants revealed two separate parameter domains. A consequent hypothesis that the node degree distribution could follow the universal double exponential law was successfully verified by selected model comparison using the AIC criterion. BIND and DIP data for H. pylori, E. coli, S. cerevisiae, D. melanogaster, C. elegans and A. thaliana were used for this purpose. A linear change in the magnitude of the distribution components with proteome size was observed, manifesting the evolutional stability of the process of developing the protein interaction network. Proposed kinetic model of protein evolution, considering the two hypothetical protein classes, first, with a relatively rapid emerging rate and a short characteristic residence time, and the second one, with the opposite properties, analytically described the nature of bi-exponential pattern. The model presents a situation in which evolutionary conserved proteins increase their interactions due to specific kinetic conditions. Thus, we oppose the opinion that the majority of such interactions are biologically significant, and, therefore the older parts of interactome are more complex. We believe that our interactome results support the hypothesis of Stuart Kaufman, presented in his book "The Origin of Order", that random mutations and natural selection constitute the origin of order and complexity.
Keywords: Protein interaction network, Exponential model, Scale free network, Power law
The degree of a node (or connectivity) is the number of edges that are adjacent to it. From the theoretical point of view, it is one of the basic measures characterizing the importance of the node in the network. Although the power law (PL) and the generalized power law supplemented with an exponential cut-off (GPL-EC) were widely popularized (Wagner, 2001; Jeong et al.. 2001) as the rules describing the distribution of the node degrees in proteinprotein interaction network, attempts at a more exact mathematical description are still being undertaken (Thomas et al.. 2003; Berg et al.. 2004). The reasons are both of practical and methodological nature. The first reason pertains to the still evolving databases, and the second one concerns the facts that the usually simple shape of arrangement of experimental points may be fitted in various manners giving at different theoretical assumptions quite similar results. According to the DIP data (see Materials and Methods) we could observe that the degree distribution of nodes of S. cerevisiae protein interaction network follows approximately a PL or a GPL-EC, but only for the degree values smaller than 10. For higher values of we saw a serious discrepancy between the theory and the experiment, already reported by others as an exponential decay (Wilhelm et al.. 2003).
There are additional indications (Barabási and Oltvai, 2004; PereiraLeal et al.. 2005) that the biological network characteristics may contain an exponential component. The main aim of the present paper is to resolve whether by using a more complex exponentialtype model one can better describe the distribution of node degree in the protein interaction network. Developing the above idea we proposed to consider a node distribution as a linear combination of exponential decays Ai exp (−λi k ) , with amplitudes Ai and decay constants λi being positive values. Our method applied to S. cerevisiae DIP data revealed two separate domains of λi , with two characteristic values of the parameters related to the relatively "fast", then "slow", tendency of a distribution to decay along k-axis. This led to the natural concept that a double exponential curve a1 exp(-d1k) + a2 exp(-d2 k) could be a better model of the node degree distribution than the standard or modified power law. This supposition was confirmed by using BIND or DIP data for 6 different organisms and the AIC criterion (see Materials and Methods). The obtained results led to analysis of the dependence of both exponential contributions to the total protein pool on proteome size, clearly indicating a linear trend. In consequence, this analysis helps us to better characterise the evolutionary mechanism leading to the observed double exponential distribution and points out its universal elements.
To explain the bi-exponential character of node degree distribution, the kinetic model of protein network evolution was proposed. It relates the searched distribution formula to the parameters describing the rate of some creation and disruption processes, postulated as being important in formation of the net. According to our model, two basic types of proteins, marked "1" and "2", with a different dynamics of evolutional behaviour were assumed. They were shown to be good candidates, from a statistical point of view, for the low-connected nodes and hubs, respectively.
The discussed results suggest that the process of evolution leads to a "biological" order in the interactome. Therefore, they support the hypothesis of Kaufman, (1993) that the process of random mutation and selection always leads to complexity.
Protein interaction network data for H. pylori ( = 724 nodes, Ne = 1403 edges) and S. cerevisiae (analogous values 4135 and 7839) were taken from Coevolution and Self-organisation in Dynamical Networks data sets (COSIN, http://www.cosin.org) derived from the Database of Interacting Proteins (DIP, http://dip.doe-mbi.u cla.edu/). Data for E. coli (399 and 312), D. melanogaster (7910 and 23128), C. elegans (3227 and 5026) and A. thaliana (487 and 959) were taken from Biomolecular Interaction Network Database (BIND, http://www.bind.ca/Action). Only single protein-protein interaction records (without self-interaction) were analyzed. No noninteracting proteins were reported.
According to our method of linear combination of exponential decays (LCED), a S. cerevisiae node degree distribution (histogram) was tentatively described by the sum:
i_max
nK = ∑ Ai exp(-λiK) (1)
i=0
where nk was a number of k -degree nodes and i _ max was the maximal value of a sum index i .Equation 1 was fitted to the experimental data, at _ max = 50 and gridded spectrum of decay constants λi = {0, 0.025, 0.050, 0.075… 1.250}. The fit had been repeated 20 times to find the sets of amplitudes Ai , and then the respective averages
In the final modelling with a double exponential law (DEL),
nK =a1 exp(-d1K) + a2 exp(-d2k) (2)
In the alternative modelling with a PL,
nK = Ak−γ (3)
and with a GPL-C,
nK = A (K+ K0)−γ exp(-K/Kc) (4)
The fits were performed in the range 1 ≤ k ≤ 15, using NRP once (without squared substitution of amplitude), and at default starting conditions (1.0).
To rate the quality of the proposed models, corrected Akaike's Information Criterion (AICc) was adopted, defined as:
AICc = Z ln( σ2 ) + 2m + 2m(m + 1) / z- m -1 (5)
where σ 2 is the average squared residual for a given model, - the number of model m parameters, and .- the number of observations (Burnham and Anderson 2004). In the case of PL, m = 2. For GPL-EC and DEL, m = 4. The number of analysed points was z = 15 in each competing model. Models with a smaller AICc value were being favoured.
In the theoretical considerations, the total proteome size ( * NP )of the analysed species was assumed to be equal to the number of open reading frames, i.e., 1788 for H. pylori, 4285 for E. coli, 6307 for S. cerevisiae, 14218 for D. melanogaster and 18944 for C. elegans (Liu and Rost, 2001) or 28952 for family members of A. thaliana (Horan et al., 2005). Due to division by the scaling factor , where:
SC = n0 + nK >0 / N*P (6)
describes the ratio of the extrapolated size of the analysed probe to the size of the total proteome, the DEL model amplitudes for accessed data, a1 and a2, were transformed into hypothetical values a1*= a1 / sc and a2*= a2 / sc , for the total species proteome (see Appendix 1). In eq. 6 the unknown value n0 was replaced by a1 + a2 . Then, the expected amount of proteins in considered contributions 1 and 2 to the total proteome was estimated by the sum of infinite geometrical series
∞
Ni* = ∑ ai*exp( -diK ) i = 1, 2 (7)
K=0
leading to:
N1* = a1* / 1 - exp( -d1 ) (8)
N2* = a2* / 1 - exp( -d2 ) (9)
In the estimation of the parameters of the model of protein network evolution (Appendix 2) eqs. A.2.8-11 were applied.
It was observed that the distribution histogram of node degree of S. cerevisiae protein-protein interaction network exhibits a well-ordered pattern in the range1≤ k ≤ 25 (Fig. 1).

Figure 1: The distribution histogram (nk) of node degree (k) of S. cerevisiae protein-protein interaction network. Presented fits are: the upper line - a power law (PL): 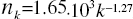 the bottom line - a generalized power law supplemented with an exponential cut-off (GPL-EC):
the bottom line - a generalized power law supplemented with an exponential cut-off (GPL-EC): 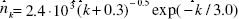 Zero values are not shown.
Zero values are not shown.
Above that range statistical fluctuations prevailed and quantization perturbed the continuity of analysed characteristics of the network. Attempts to describe the investigated distribution by a PL: A=(1.65 ± 0.02).103, γ = (1.27 ± 0.02)(upper line), or by a GPL-EC: A = (2.4 ± 0.7).103,k0 = (0.3 ± 0.7),γ =(0.5 ± 0.3),kc =(3.0 ± 0.4)(bottom line) gave good results only in the range 1 ≤ k ≤ 10.The PL parameters obtained,γ = 1.27 and A/Np = 0.40, are consistent with γ = 1.32 and A/Np = 0.42 for the whole yeast interaction network (Yu et al.. 2004). A different picture is seen in case of the GPL-EC model. One can notice a big discrepancy between our result and those for a Mean standard error (s.e.) is also presented. small sample of 1870 nodes (Pastor-Satorras et al., 2003), which may indicate the narrow area of applicability of the cut-off formula.
The proposed LCED method (Fig. 2) revealed two narrow ranges of decay constants spectrum with dominant amplitudes at λ7 = 0.175 and λ25 = 0.625 (characteristic values of node degree: 1/ λ7 = 5.7, 1/ λ 25 = 1.6). Half-width of the observed peaks equals 0.025 and 0.050, respectively. An example of one in 20 fits performed to obtain the above spectrum is also presented (Fig. 3). As it is seen here, and in the case of other fits (data not shown), their qualities, especially in the range of values k > 10, are better than the estimation with standard or modified power law.

Figure 2: Linear combination of exponential decays method (LCED) applied to the data for S. cerevisiae (Fig.1). Two regions of decay constants (λ) spectrum with dominant amplitudes Ai at 7 =0.175 and 25 = 0.625 are clearly seen. Shown values are averages of adequate amplitudes of 20 multi-exponential fits mean standard error (s.e.) is also presented.

Figure 3: An example of one of 20 fits to the experimental data (Fig. 1) performed to obtain the decay constants spectrum (Fig.2) The nK is the number of k-degree nodes. For clarity, the open circles denote group averages. Zero values are not shown.
As a result of the above, it was hypothesized that our combination, even reduced to a double exponential formula, could provide a better description of the node degree distribution than the considered power law type models. The examples of yeast and five other species were analysed for k < 15. Corresponding fits of proposed DEL models are presented in (Fig. 4) a-f and Table 1.Their qualities are confirmed by AICc values, which favour bi-exponential approximation in 5/6 of the investigated cases (Table 2). Plots of alternative fits are not shown.

Figure 4: The distribution histogram (nk) of node degree (k) for different species. Continuous line is the fit of a double exponential law (DEL). Parameters of the DEL models are presented in Table 1.
A. Helicobacter pylori.
B. Escherichia coli.
C. Saccharomyces cerevisiae.
D. Drosophila melanogaster.
E. Caenorhabditis elegans.
F. Arabidopsis thaliana.
| a1 | a2 | d1 | d2 | |
|---|---|---|---|---|
| H. pylori | 507.409 | 44.529 | 0.743 | 0.157 |
| E. coli | 1166.020 | 219.041 | 1.898 | 0.762 |
| S. cerevisiae | 2592.380 | 197.464 | 0.616 | 0.170 |
| D. melanogaster | 5783.780 | 837.777 | 1.005 | 0.187 |
| C. elegans | 7307.120 | 389.915 | 1.564 | 0.278 |
| A. thaliana | 486.548 | 68.659 | 1.234 | 0.220 |
Table 1: Parameters of the fitted DEL models.
Some parameters of DEL models vary with proteome size. The size N1* and N2* of distinguished protein groups increases with the total number of proteins N*P (Fig. 5). There was no detected essential dependence of decay constant d1 and d2 on the proteome size.

Figure 5: A-B The variation in the estimated number of proteins 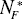 and
and 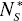 of a given protein class with proteome size
of a given protein class with proteome size 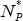 . The following data points represent: (from left) H. pylori, E. coli, S. cerevisiae, D. melanogaster, C. elegans and A. thaliana. A. Protein class F. B. Protein class S. Continuous line - linear trend.
. The following data points represent: (from left) H. pylori, E. coli, S. cerevisiae, D. melanogaster, C. elegans and A. thaliana. A. Protein class F. B. Protein class S. Continuous line - linear trend.
The results presented above confirm recent reports (Goldberg et al., 2005) suggesting the "break" of a power law in the global description of the protein interaction network. Actually, we can suggest that this "break" may be caused by the second exponential term in node degree distribution, which does not affect strongly the formula in the range of the node degree smaller than 10, but may be essential elsewhere.
Initial inspection of the data shown in Fig. 1 reveals that GPL-EC, the 4-parameter improvement of PL (bottom line), fits better than PL alone (upper line), but is still a very long way from perfect. Hence we decided to introduce a more general description.
In accordance with our idea, protein interaction network consists of subpopulations of vertexes described by a similar statistical formula, but with different parameters. As a universal formula we choose exponential decay, which is consistent with the suggested model of network evolution (see Appendix 2).
The proposed LCED method revealed the spectrum of decay constants and the magnitude of subpopulational contributions into the degree global distribution (Fig. 2). Two classes of nodes with the values of decay constant lying closely together were clearly distinguished. Good quality of fits (Fig. 3) testifies to the utility of the method and the acceptance of the formula.
Reducing the huge number of parameters of a general model and taking into account the above observation, we propose to limit the number of decay components to only two items, indexed by 1 and 2. It did not weaken the fitting abilities for different species in the range 1 ≤ k ≤ 15 (Fig. 4a-f, Table 1), which was confirmed by the AICc criterion. As seen in Table 2, the DEL models are the best in 5/6 of investigated cases and just a little worse (2nd place) than the winner in one case. Generally, they are more effective for networks with big proteomes (the PL model for a small probe of A. thaliana may be an exception) than for sets with a small protein number; PL or GPL-EC models may give similar results
| DEL | PL | GPL-EC | |
|---|---|---|---|
| H. pylori | 56.4 [1] | 73.2 [3] | 58.3 [2] |
| E. coli | 6.0 [1] | 43.0 [3] | 6.4 [2] |
| S. cerevisiae | 94.6 [1] | 135.8 [3] | 112.4 [2] |
| D. melanogaster | 97.4 [1] | 122.1 [3] | 109.4 [2] |
| C. elegans | 52.5 [1] | 66.5 [2] | 90.9 [3] |
| A. thaliana | 38.9 [2] | 37.2 [1] | 130.5 [3] |
1AICc criterion numbers are shown and the adequate ranks are given in parenthesises.
Table 2: AICc ranking of the models1.
Documented changes in the dimensions of the indexed protein classes with proteome size (Fig. 5) indicate a similar tendency for linear increase for the first (a) and the second (b) component of proteome. This way the ratio N1*/ N2* ≈ 2 .5 seems to be a universal constant for a wide group of organisms. The contribution of each class of proteins to the summary distribution was shown in the example of a yeast probe (Fig. 6).

Figure 6: The contribution of "F" and "S" protein class to the overall distribution. The insets 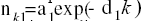 and
and 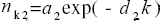 were plotted (continuous lines 1 and 2) for the parameters of S. cerevisiae (Table 1). The broken line represents fitted summary distribution
were plotted (continuous lines 1 and 2) for the parameters of S. cerevisiae (Table 1). The broken line represents fitted summary distribution 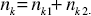
As seen for small values of k , the two classes contribute to the global distribution. For k > 10, the first class vanishes and the second class clearly dominates. The latter class may be related to so called hubs. It is worth stressing that the second class of proteins may bear only a few links, too.
It seems that the proposed double-exponential model is a simplification of a hypothetical multi-componential model describing the full spectrum of contributions from different classes of proteins. The analysed data indicate that there probably exists the third, small amplitude class of yeast proteins (not visible in Fig. 2), which may be related to the "super" hubs connecting hundreds of nodes; however, a "false positive" error cannot be excluded.
Although the two protein classes clearly dominate, the analysed subpopulations do not form spikes along the decay constant axes, but have some definite width. We believe that more sophisticated analysis of discussed contributions, considering their continuous representation, should fully describe protein network statistics and reveal new properties of the proteome system.
As mentioned beforehand, to specify our hypothesis, we proposed a simple mathematical model of protein network evolution (Appendix 2). The applied assumptions permit duplication events to occur even more often than the appearance of "new" types of protein encoding genes. Such behaviour is suggested by the observation that gene-copy number within a family is often changed during the process of speciation (Cheng et al., 2005; Ma and Gustafson, 2005; Ting et al., 2004). However, to avoid an enormous expansion of the system, we assumed that the speciation processes are no more frequent than deletion episodes effectively leading to the elimination of proteins. On the other hand, one can detect evolutionary conservation of genes present even in different kingdoms. Therefore, the probability of multiplication of old "proteins" is similar to the probability of multiplication of"young" proteins in a given genome. The facts mentioned above were "silently" included in the model. It relates amplitudes and decay constants to the emergence rates, q1 and q1 , effective elimination rates, γ1 and γ2 , and interaction gaining rates, ν1 and ν2 of the two classes of proteins, with different dynamics of evolutional performance. This difference in dynamics of the evolution of proteins manifests in the observed difference between"fast" and "slow" tendency in the variation of the node degree distribution along k-axis. In general the above parameters may differ for different evolutional pathways.
According our model, the linear trend in Fig. 5 may be related to the stable dynamics of evolution of investigated classes of proteins during the inter space progress. Indeed, with equations A.2.12-14 it is easy to show that the observed dependence calls for stability of the ratio. q1 γ2 / q 2 γ1 This linear trend also suggests that for the total proteomes the corresponding amplitudes of calculated probability (frequency) of the occurrence of a node with a given degree may remain approximately constant. In a sense, we showed not a scale-free distribution but a scale-free evolution.
As the analysed decay constants d1 and d2 do not exhibit a clear tendency to change, we may simply imagine that during evolution γ1 ,γ2 , ν1 and ν2 remain approximately constant (see eqs. A.2.10-11). According to this picture, q1 and q2 slowly evolve in a stable manner ( q1 / q2 = const ), governed, for example, by the varying amount of DNA, which accounts for the change in the global protein pool (see eq. A.2.12).
To make our considerations more quantitative we estimated values q1 , q2 , γ1 and γ2 , assuming that ν1 =ν2 =0.1 [1/mln years] (Berg et al., 2004). It is seen (Table 3) that first class of proteins may be characterized by a relatively rapid emerging rate q1 and also relatively rapid elimination γ1 rate (or short characteristic residence time) when to compare with the second class of proteins.
The proposed mathematical model of evolution suggests unexpected explanation of the observation of Barabasi and co-workers that more densely interconnected parts, "motives" of the interaction network, are more strictly evolutionary conserved (Wuchty et al., 2003). Intuitively, one can suppose that proteins belonging to such motives are evolutionary conserved because they are required for maintaining the connections in such motives. But the results of our simulations suggest an exactly opposite explanation: the old proteins (evolutionary conserved proteins) are more interconnected because they are simply old enough. This explanation although surprising for us, does in fact have sense. Since the majority of the proteins are not interacting (for example, protein-protein interaction network of yeast contains only approximately 30000 protein-protein interactions (according to the estimation of Kumar and Snyder, 2000) and more than 36000000 protein-protein pairs), and the protein interaction network is evolutionary conserved (see, for example, Matthews et al., 2001), it is likely that the majority of interactions have biological significance and that interactions appear gradually during the process of evolution. It is also likely that new "proteins" have no interactions or have a small number of interactions. During the process of evolution these proteins slowly gain new "useful" interactions. If they belong to the class 2, they may even gain many such interactions. This process leads to a well-ordered protein-protein interaction network in which proteins are not randomly connected and in which one can distinguish "modules" of interacting proteins.
As we have already referred to in the Introduction, our results support the hypothesis of Stuart Kaufman that natural selection, random mutations and the process of evolution are the source of order in biological systems. This paper shows a random process of evolution leading to complex and non-random systems. Although it remains an open question whether the random process is rapid enough to lead to creation of structures as complex as multi-enzymatic complexes or flagelles, we believe that a right step in the proper direction has been taken.
We would like to thank Doktor Christophe Pakleza for his help in the application acknowledgement of Mathematica to numerical calculations. Fianancial support from the ministry of Science and Higher Education geant PBZ-MNil_2/1/2005 and postdoctoral fellowship from the foundation for polish Science to S.K are gratefully acknowledged.
It was assumed that each data set analysed is only a homogenous part of the total proteome of a given species. Then the fitted DEL model formula and the hypothetical distribution of the total population of proteins of a given organism (see Appendix 2) are related in the proportion:
nK def a1 exp( -d1k ) + a2 exp ( -d2k ) NP
= = (A1.1)
nk* a1* exp( -d1k ) + a2* exp (-d2k) Np*
where a1* and a2* are the amplitudes of a hypothetical distribution for the total population, Np is the extrapolated size of the analysed probe and Np* is the total size of proteome.
In the above ratio, Np value includes interacting proteins ( Nk>0 ) and also non-interacting ones ( n0 ) - not included in the investigated data sets, so that:
Np = n0 + Nk>0 (A.1.2)
As eq. A.1.1 is fulfilled for each node degree and for different decay constants d1 and d2 , it should be:
a1* = a1 / sc (A.1.3)
a2* = a2 / sc (A.1.4)
where the scaling factor equals to:
n0 + Nk>0
sc = (A.1.5)
Np*
Let us consider protein interaction network containing two classes of proteins (namely 1 and 2) characterized by different dynamics of evolutional performance, i.e., emerging with the rates q1 and q2 (as non-interacting at the beginning), then gaining some interactions with the rates ν1 and ν2 , and being eliminated with the rates γ1 and γ2 - per protein. All mentioned rates are assumed as being distinct and constant.
A number of selected proteins of a given class δNi*(i=1,2), originated within small period of time , vanishes with age a according the equation
dδNi*
= - γiΔ Ni* i = 1, 2 (A.2.1)
da
with an initial condition
Ni*|a=0 = qiδt i= 1, 2 (A.2.2)
The resolution of eqs. A.2.1 and A.2.2 represents the exponentially diminishing course
δNi* = qiδt exp( - γia) i= 1, 2 (A.2.3)
The assumed continuous approximation and linear increase in protein connectivity
K = νia (A.2.4)
and also the relationship , let us to transform eq. A.2.3 into the formula
qi γi
δNi*= δK exp( - K ) i=1, 2 (A.2.5)
νi νi
which integrated within successive intervals [k, k+1] indicates the number of k-degree proteins of class "i" , , equal to
qi γi γi
nki* = (1 - exp( - ) ) exp( - K ) i=1, 2 (A.2.6)
γi νi νi
Now, the total distribution of node degree, nk*, where nk* = nk1* + nk2*, may be written in the double-exponential form:
nk* = a1* exp( -d1k ) + a2* exp( -d2k ) (A.2.7)
The symbols introduced above mean
q1 γ1
a1* = (1 - exp( - ) ) (A.2.8)
γ1 ν1
q2 γ2
a2* = (1 - exp( - ) ) (A.2.9)
γ2 ν2
γ1
d1 = (A.2.10)
ν1
γ2
d2 = (A.2.11)
ν2
A contribution of "i " class proteins in eqs. A.2.7 formally vanishes for K > ζevi −1, where is the time of evolution of interactome. Thus the index k should not exceed max[ ζev1 −1, ζev2 −1 ] Assuming a relatively high value ζe ( >> 1/ νi ), by summation of a superposition of geometrical series nk*described by the eq. A.2.7 over 0 ≤ k ≤ ∞ , one can obtain the total size of proteome : Np*
q1 q 2
Np* = + (A.2.12)
γ1 γ2
with a distinguished levels of class contribution
q1
N1* = (A.2.13)
γ1
and
q2
N2* = (A.2.14)
Y2