Journal of Antivirals & Antiretrovirals
Open Access
ISSN: 1948-5964
ISSN: 1948-5964
Research Article - (2016) Volume 0, Issue 0
Three influenza A virus isolates, one from a human, one from a chicken, and one from a wild bird dropping, were used to infect human peripheral blood mononuclear cells (PBMCs) in culture. The influenza strains were H1N, H5/H7-N1/N2 (mixture) and H9N, respectively and were assessed in vitro in human PBMCs in the presence of Phytohemagglutinin (PHA). Viral replication was estimated by visual cytopathic effect (CPE). H1N2 CPE included budding of lymphocytes, fusion of infected cells with neighboring lymphocytes, and syncytial formation. H5/H7- N1/N2 and H9N2 each caused CPE that included bulging of cells with large vacuoles. The supernatant of infected lymphocyte culture was used to infect MDCK cell lines. Influenza viral RNA was detected in extracts of MDCK cell lines and from lymphocytes infected with each of the three virus strains as judged by RT-PCR, confirming that all three isolates were able to replicate in human lymphocytes in vitro. The antiviral activity of a mixture of honey, ginger, and garlic (HGG) extracts, an over the counter drug commonly used in Pakistan to treat patients with influenza virus, was compared to the antiviral drug amantadine for its ability to decrease replication of the H1N2 strain in human PBMCs in vitro. HGG significantly inhibited H1N2 virus growth as judged by CPE, haemagglutination assays, and qRT-PCR. Interestingly, HGG also appeared to promote proliferation of human lymphocytes. These finding suggest that a mixture of crude extracts from honey, ginger, and garlic may be clinically useful against influenza virus.
<Keywords: Influenza Virus; Lymphocytes; Honey; Ginger; Garlic; Haemagglutination
Influenza viruses are members of the Orthomyxoviridae family. They have a segmented, linear, single-stranded, negative sense RNA genome in an enveloped virion [1]. On the basis of antigenic properties of their matrix or nucleoproteins, influenza viruses are categorized as influenza A, B or C. Widespread outbreaks in humans are caused by type A and B viruses [2,3].
Influenza virus occurs globally and is a major cause of respiratory tract infections, with an annual attack rate estimated at 5%–10% in adults and 20%–30% in children. Illnesses can result in hospitalization and death, mainly among high-risk groups (the very young, elderly or chronically ill). Worldwide influenza virus is reported to cause 3 to 5 million severe clinical infections and 250,000 to 500,000 fatal cases annually [4,5].
Host innate and adaptive immune responses against influenza virus play an important role in viral clearance and protection [6]. The innate immune system provides a rapid first line of defense against influenza virus infection. In addition, dendritic cells (DCs) reside in the upper respiratory tract epithelia, the site of entry for Influenza virus. DCs are professional antigen-presenting cells (APCs) and they rapidly mobilize to present influenza virus-derived antigens to T cells [7-10] to induce an adaptive immune response that includes neutralizing antibodies and virus-specific CD8+ cytotoxic T cells and CD4+ T helper cells [11]. T cells are mainly directed to conserved proteins of influenza virus and therefore display cross-reactivity against a variety of different subtypes of influenza virus. T cell mediated immunity thus contributes to socalled hetero-subtypic immunity and protects against antigenically distinct, potentially pandemic influenza viruses [6].
Regardless of the underlying immune mechanisms that protect against various strains of influenza virus, only limited information is available on the direct interaction between influenza virus strains and host T cells [12,13]. Studies with highly virulent influenza A virus strains resulted in the destruction of lymphocytes (e.g. RP9 avian lymphocytes cell line) and histopathological necrosis of lymphoid tissues [12,14,15]. However, whether influenza virus is able to propagate in human lymphocytes remains to be fully elucidated. In the present study three recently isolated influenza viral strains were used. H1N2 was isolated from a human nasal swab from a woman who had just returned to Karachi from Turkey. H5/H7-N1/N2 mixture was isolated from a bird dropping sample collected at Karachi University, a junction point for stray and migratory birds. H9N2 was isolated from the lung of a poultry bird (chicken) from a poultry farm in Karachi. We report here that: (i) influenza virus strains can grow in human lymphocytes; (ii) the H1N2 strain resulted in budding of lymphocytes followed by fusion and syncytial formation. The H5/H7-N1/N2 mixture and H9N2 produced bulging of cells with massive lymphocyte vacuolation and, (iii) a significant in vitro antiviral effect of a honey, ginger, and garlic extract was seen against influenza virus in human lymphocytes in tissue culture. For centuries, this combination of honey, garlic, and ginger has been used as a traditional medicine because of the presumed therapeutic properties of each, including antibacterial, antifungal, and antiviral activity [16-19]. So far, the antiviral drug amantadine, M2 ion channel inhibitor, has been used most commonly for the treatment of influenza virus infections worldwide. Therefore, we used amantadine as the positive control for the influenza virus inhibition assay. HGG significantly inhibited influenza virus growth in vitro in human lymphocytes, suggesting that this crude extract may have effective anti-influenza therapeutic activity in vivo .
Viruses
The influenza strains included H1N2, isolated from a human nasal swab. H9N2 isolated from a lung sample of a commercial poultry bird (chicken), from the Poultry Hawker in open poultry markets, situated in Abu-al-Asfahani Road, Gulshan - e Iqbal, Karachi (nearby Karachi University), geographic coordinates are 24° 58' 05" North, 67° 06' 18" East. H5/H7-N1/N2 (mixture), isolated from a bird dropping sample from stray bird collected from the underlay grounds at Karachi University. This area is heavily bird nested due to huge trees of Azadirachta Indica (Neem) and Ficus Religiosa (Peepal). Karachi University is a junction point for stray and migratory birds; geographic coordinates are 24° 56' 08" North, 67° 07' 24" East. The influenza viruses were propagated in embryonated eggs and identified by ELISA and RT-PCR. The alantoic fluid was aliquoted and stored at -80°C as virus stocks. Embryonated eggs were bought by Sindh Poultry Vaccine Centre (SPVC), Karachi, Pakistan. No animal was involved directly during sample collection/isolation.
Herbs and drugs
Honey, ginger, and garlic mixture (HGG) was purchased from a local market in Karachi. This mixture is sold locally as a treatment for the flu. HGG was dissolved in RPMI 1640 tissue culture media at 5% (V/V). 10 fold serially dilutions were made using the same tissue culture media to a final concentration of 0.005% and then filter sterilized. Amantadine (Amantadine hydrochloride capsule) was purchased from Novartis (Pakistan).
Propagation of influenza viruses in human lymphocytes
All subjects were enrolled at the University of Karachi, Pakistan, under Ethical Committee of Karachi University, Pakistan. Written informed consent was received from all participants prior to inclusion in the study. Lymphocytes were isolated from 20 ml of blood from healthy O+ individuals. Written informed consent forms were received from all participants who donated blood. Blood was collected and immediately transferred to EDTA containing purple caped tubes. In 15 ml Falcon tubes, 5 ml of RPMI-1640 culture medium (without FBS) and equal volumes of blood were pipetted. 10 ml histopaque was pipetted in another 50 ml Falcon tube and the diluted blood in RPMI-1640 was layered on top. Tubes were spun at 400 x g for 20 minutes at room temperature in a centrifuge with swinging buckets. PBMCs appeared at the junction of the two layers. The upper layer was discarded and the PBMCs (buffy layer) were transferred to another sterile tube. The PBMCs were washed with RPMI-1640 medium (without FBS) and spun at 300 × g for 20 minutes at 4°C. The pellet was re-suspended in 1 ml RPMI-1640 (10% FBS). Lymphocytes were seeded in 24 wells plate at a concentration of 1 x 106 cells/cm2 in RPMI-1640 medium containing 5% v/v FBS, 5 μg/ml PHA and 10 mg/ml of penicillin and streptomycin. These cells are non adherent. Cells were infected ~48 h. later when the lymphocyte suspension was confluent. Prior to viral inoculation, the medium containing the suspended cells was harvested from each well, centrifuged at 200 × g for 5 minutes and the supernatant discarded. The cell pellets were washed once with phosphate buffered saline (PBS), then 100 μl of incomplete RPMI-1640 was added and the cells transferred back to tissue culture wells. The cells were infected with H1N2, H5/H7, N1/N2 and H9N2 viruses separately and the viruses were allowed to adsorb at 37°C for 60 minutes in a 5% CO2 humidified incubator. The cell suspension was harvested, centrifuged at 200 × g for 5 minutes, the supernatant discarded and the cells were washed once with PBS. 100 μl of incomplete RPMI-1640 was added and the cells were transferred back to tissue culture wells. The infected cells were incubated at 37°C and 5% CO2 for 24 hours. After 24 hours, cells were observed under a microscope for cytopathic effect (CPE). Afterwards supernatants were collected and 100 μL of each was used to infect the MDCK and incubated at cells at 37°C and 5% CO2 for 24 hours in order to confirm the propagation of influenza virus in the lymphocyte. After 24 hours, MDCK cells were observed under a microscope for cytopathic effect (CPE) and supernatant along with cells and cell debris were collected. The RNA is extracted from the remaining supernatant, cells and cell debris of the infected lymphocytes and infected MDCK cells (as a control). Finally, relative viral propagation was estimated by RT-PCR.
Antiviral effect of HGG on H1N2 in human lymphocytes estimated by inhibition of viral induced cytopathic effect (CPE)
Lymphocytes were prepared as above and infected with influenza viral strain H1N2. The infection medium was RPMI-1640 containing 10 mg/ml penicillin/ streptomycin, 4 μg/ml trypsin and 1 μg/ml EDTA. Following virus adsorption and removal of excess virus by washing cells, the infected cells were fed with RPMI-1640 tissue culture medium containing HGG or Amantadine. Positive control wells were infected with H1N2 virus without HGG or Amantadine. Negative controls included uninfected cells and uninfected allantoic fluid. The lymphocytes were incubated at 37°C and 5% CO2 for 24 h. Relative virus replication was estimated by observation of virus induced CPE of the lymphocytes. In addition, total cell extracts were prepared to estimate relative virus replication by hemagglutination assays for virions and following RNA isolation, by quantitative real time PCR (qRT-PCR) for viral mRNAs, as described below.
Hemagglutination inhibition assay to estimate virus replication
Influenza viruses are characterized by their ability to agglutinate RBCs. This hemagglutination activity can be used to estimate the amount of virus present. Viral dilutions from cells grown in the presence of HGG or Amantadine were mixed with chicken RBCs in Ushaped 96 well microtiter plates. Chicken RBCs were supplemented with 1.6% sodium citrate (Sigma, USA) in sterile water, separated by centrifugation (800 × g, 10 min, room temperature) and washed three times with sterile PBS. Serial dilutions of viral supernatant in 30 μl were added to wells of 96-well U-shaped micro-plates. 30 μl of a freshly prepared 1% suspension of RBCs were then added to each well and the plates were incubated for 30 minutes at 37°C. The presence or absence of hemagglutination was observed in each well. In the absence of virus there is no hemagglutination and the RBCs fall to the bottom of the U shaped well and form a distinct button or pellet. When there is sufficient virus in the virus dilution to produce extensive hemagglutination, the virus and RBCs form a lattice-work web and no RBC button is seen.
Quantitative real-time PCR analysis to estimate virus replication
RNA Extraction
Total cellular and viral RNA was extracted by mixing 750 μl TRI Soln with 250 μl of infected lymphocyte suspension, and incubating at room temperature for 5 minutes. 150 μl of chloroform was added, the samples were vortexed, and incubated at room temperature for 2-3 minutes. The RNA samples were spun at 14000 × g for 15 minutes at 4°C and the aqueous layer was transferred to a new eppendorf microcentrifuge tube. 375 μl of isopropanol was added, the samples were vortexed for 5 seconds and incubated for 10 minutes at 4°C. The samples were spun at 14000 × g for 10 minutes and the RNA pellet was washed once with 1 ml of 75% ethanol (chilled) and the samples were spun at 12000 × g for 10 minutes. The pellet was air dried and resuspended in 100 μl of DEPC water. The RNA concentration was estimated by nanodrop.
Primer selection
Two sets of primers that specifically amplify the Matrix mRNA of influenza A virus and human GAPDH mRNA were used [19]. M sequence forward: 5´AGA TGA GTC TTC TAA CCG AGG TCG3´; reverse: 5´TGC AAA AAC ATC TTC AAG TCT CTG3´. GAPDH sequence forward: 5´AGA TGA GTC TTC TAA CCG AGG TCG3´; reverse: 5´TGC AAA AAC ATC TTC AAG TCT CTG3´ These primers were synthesized by Integrated DNA Technologies (lowa). Primers were aliquoted at a final concentration of 1 nmol/μl and stored at -20°C.
cDNA synthesis
First-strand cDNA was synthesized using Revert-Aid First Strand cDNA Synthesis kit (Thermo Scientific Inc., USA) using oligo(dT)18 primer provided with the kit. RT-PCR was done using 0.5 μl Promega Taq polymerase (Promega, WI). The reaction mixture contained 5 μl of 5 Master Mix (Promega, WI), 1.5 μl of 25 nM MgCl2 (Promega, WI), 2 μl of 12.5 nM dNTPs (Applied Bio-system, NY), 20 pmol each forward and reverse primer, 2 μl cDNA template, 12 μl DNAse/RNAse free water in a 25 μl reaction volume.
Preparation of GAPDH standard
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) from human lymphocyte cells mRNA was reverse transcribed, amplified and the presence of a single PCR product was verified on a 2% agarose gel. The amplified product was than purified using Wizard SV Gel and PCR Clean-up kit (Promega Corp., Madison, WI, USA) as described by the manufacturers. Briefly, 90 μl of the amplified PCR product was mixed with 90 μl of membrane binding solution, transferred to the micro column, incubated at room temperature for 10 minutes, spun at 18000 × g for 1 minute, and the flow through was discarded.
The PCR product in the spin column was washed using 700 μl washing buffer and the samples were spun at 18000 × g for 1 minute. The flow through was discarded and the PCR product in the spin column was re-washed by adding 500 μl of washing buffer and the samples were spun at 18000 × g for 5 minutes. The settled liquid in the collection tube was discarded and the samples were dry spun at 18000 × g for 5 minutes. The collection tube was discarded and the column was transferred to a new pre-labeled eppendorf microcentrifuge tube.
40 μl of DEPC treated water was added to each of the samples, incubated for 1 minute at room temperature, and spun at 18000 × g for 1 minute. The column was discarded and the cleaned PCR product was stored at -20°C until required. The PCR product was quantified by spectrophotometry, and copy numbers were calculated by the following formula:
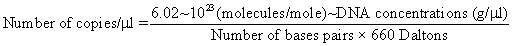
A series of duplicate 10-fold dilutions ranging from 2.5 × 106 to 2.5 × 102 copies were used as standards with every PCR run on an ABI Prism SDS 7000 (ABI, Foster City, CA, USA).
Relative quantification of viral matrix protein mRNA by Real-time PCR
PCR for viral matrix cDNA and human lymphocytes GAPDH cDNA were done to allow relative quantification of matrix mRNA as an estimate of relative viral replication. qRT-PCR was performed using the Applied Bio-systems SYBR Green PCR Core Reagents (Applied Bio-systems, NY). The reaction mixture contained 2.5 μl Master mix, 2.5 μl MgCl2, 2 μl dNTPs, 0.25 μl UNG, 0.125 μl Taq Gold, 20 pmol each of forward and reverse primer, 2 μl cDNA template and 8.625 μl DNAse/RNAse free water in a 25 μl reaction volume.
Real Time PCR was performed with the following cycling conditions: UDG pre-treatment for 10 minutes at 50°C and initial denaturation for 10 minutes at 95°C once and then 15 s at 95°C and 1 minute at 60°C, 40 times. An additional melt step was used in the Realtime PCR high resolution melting (HRM) with holds at 95°C for 15 seconds, 60°C for 1 minute, 95°C for 30 seconds and 60°C for 15 seconds.
Statistical analysis
Data were presented as mean ± SD calculated using GraphPad Prism, a computer program. The data were statistically evaluated using a t-test to compare difference between the groups. A p-value of ˂0.05 was considered significant.
Influenza virus propagates in human lymphocytes
H1N2, H5/H7 (mixture) and H9N2 were cultured in human lymphocytes and CPE for each virus was determined. H1N2 caused mostly budding of lymphocytes, fusion of infected cells with neighboring lymphocytes, and syncytial formation (Figure 1). H5/H7- N1/N2 and H9N2 each produced bulging of individual human lymphocytes with large vacuoles (Figure 1). The presence of influenza A virus was further confirmed in these infected human lymphocytes by infecting the MDCK cells with the collected supernatant from the each flu viral infected lymphocytes culture.

Figure 1: Cytopathic effects (CPE) induced by influenza virus in human Lymphocytes and MDCK cells. Cells were inoculated with 4 HA units of influenza H1N2, H5/H7 (mixture) or H9N2, and incubated for 24 h at 37°C in 5% CO2 as described in Materials & Methods. A representative example of virus induced CPE with each virus isolate is shown. Each image is representative of the CPE induced by each virus observed in triplicate in two separate experiments.
The CPE shown in (Figure 1) was determined after 24 h of the infection. H1N2 and the H5/H7-N1/N2 mixture caused tubular formation, budding of cells and syncytial formation. Whereas, CPE caused by H9N2 virus included mostly single cell vacuolation. The propagation of the flu virus in human lymphocytes was more confirmed by Reverse transcription and RT-PCR (Figure 2) using universal primers for the influenza A virus matrix gene. These results strongly suggest that each of the three influenza viruses were able to replicate in human lymphocytes (Figure 2). Interestingly, the mixture of honey, ginger and garlic appeared to stimulate the proliferation of (Figure 1).

Figure 2: (A) Agarose Gel electrophoresis of amplified matrix protein mRNA of influenza viruses cultured in human lymphocytes and MDCK cells. Human lymphocytes or MDCK cells were infected in vitro and total RNA isolated as described in Materials & Methods. cDNA was prepared using primers for the influenza viral matrix protein mRNA and RT-PCR was performed as described in Materials & Methods. Lane 1: H1N2 cultured in human lymhocytes; Lane 2: H5/H7-N1/N2 mixture cultured in human lymhocytes; Lane 3: H9N2 cultured in human lymhocytes; Lane 4: 100 bp; Lane 5: H1N2 cultured in MDCK cells; Lane 6: H5/H7- N1/N2 mixture cultured in MDCK cells; Lane 7: H9N2 cultured in MDCK cells; Lane 8: 100 bp DNA ladder (Fermentas , NY). The arrow at 100 bp shows the expected mobility of the matrix mRNA PCR product. The HA and NA serotypes of these isolates was previously determined.
HGG reduced influenza virus replication in human lymphocytes in a dose response manner
In previous studies, we found that HGG reduced influenza virus replication in MDCK cell monolayers in vitro (data not shown). Here we tested the antiviral activity of HGG on our recent H1N2 human virus isolate cultured in human lymphocytes. Human lymphocytes were infected with 4 haemagglutination (HA) units of the H1N2 virus and the virus was grown in the presence of 5%, 0.5%, 0.05%, or 0.005% HGG in the media. A dose response effect on decreasing virus replication as judged by CPE was seen (Figure 3A). The viruses were similarly grown in the presence of 5 μM, 500 nM, 50 nM, or 5 nM Amantadine (Figure 3B). Uninfected allantoic fluid was used as a negative control for the inhibition of virus replication (data not shown). Control virus showed extensive CPE in the absence of HGG and Amantadine. The maximum inhibitory effect on CPE (and hence virus replication) was seen with 5% and 0.5% HGG and with 5 μM and 500 nM Amantadine.

Figure 3: HGG inhibits influenza virus propagation in a dose response manner as judged by CPE. Human lymphocytes were infected with each of the three influenza viruses as described above and in Materials and Methods and fed with RPMI-1640 without FBS containing 10-fold serial dilutions of HGG (0.005% - 5%) or Amantadine (5 μM - 5 nM). Relative virus replication was estimated on a scale of 0 to 4 based on cytopathic effects. Control virus received no HGG or Amantadine. Each bar is representative of 6 repeats (two separate experiments done in triplicate). No error bars can be seen, because for each HGG or Amantadine dose there was no variation in the CPE score of the 6 repeats.
HGG Blocks replication of influenza virus as determined by a hemagglutination assay
To further confirm that HGG decreased viral replication in human lymphocytes, a hemagglutination assay was performed using cell extract from the experiment in the previous paragraph (Figure 4). The results indicated that HGG decreased the replication of all three influenza viral subtypes in a dose-dependent manner. Aliquots of virus grown in the presence of a low dose of HGG or Amantadine caused more hemagglutination than aliquots of virus grown in higher doses of HGG or Amantadine (Figure 4).

Figure 4: HGG decoction inhibits influenza virus propagation in a dose response manner as judged by hemagglutination assays of virus from the experiment in Figure 3. Following determination of CPE in the experiment shown in Figure 3 , total virus lysates were analyzed for their ability to cause haemagglutination of chicken RBCs as described in Materials & Methods. The amount of HGG and Amantadine are shown at the top and bottom of each panel respectively, indicates the amount of HGG or Amantadine present in the tissue culture media while the virus was grown in human lymphocytes. The rightmost lane in each panel labeled “control” i.e. virus grown without HGG or amantadine incubated with the RBCs.
HGG has antiviral activity as judged by qRT-PCR for the influenza M gene
The highest and the lowest dilutions of HGG (5% and 0.005%) and amantadine (5 μM and 5 nM) were examined. Human lymphocytes were infected and treated with HGG or Amantadine as above. At 24 h post infection, total RNA was extracted. cDNA followed by qRT-PCR as described in Materials & Methods were performed to determine the levels of intracellular influenza viral mRNA for the matrix protein. There was less matrix protein mRNA when viruses were grown in the presence of HGG or amantadine and the reduction appeared to be dose-dependent. The amount of matrix mRNA was significantly reduced in both 5% and 0.005 HGG -grown virus-infected cells compared to untreated virus-infected cells (Figure 5; p = 0.0038 and 0.0105 respectively). The higher dose of HGG (5%) appeared to decrease the influenza viral matrix protein mRNA levels more than the lower dose HGG treatment. 5 μM and 0.005 μM amantadine treatment also both significantly decreased matrix mRNA expressions with larger decreases being seen at the higher drug dose (Figure 5). These results confirm that HGG can block influenza virus replication in a dose response manner in human lymphocytes in vitro .

Figure 5: Real-time reverse transcription-PCR of influenza viral Matrix (M) RNA levels normalized to GAPDH. Human lymphocytes were infected with H1N2 virus and grown in the indicated concentrations of HGG or Amantadine as described above. Total RNA was isolated 24 h post infection as described in Materials & Methods. Reverse transcription was performed to convert mRNAs to cDNAs and qRT-PCR was performed using primers specific for the viral matrix or cellular GAPDH nucleic acid sequence as described in Materials & Methods. The level of the viral mRNA was normalized to GAPDH. Standard curves based on Ct values of tenfold dilutions of template DNA were used to estimate the amount of final products, and the results were plotted as a bar graph. Data was analyzed using the GraphPad Prism computer program. One way t-tests were performed using 4 independent experiments (mean ± SD). P<0.05 is considered significantly different.
The study was preliminarily designed to investigate the interaction between influenza virus and human peripheral blood mononuclear lymphocytes (PBMCs), specifically T lymphocytes from healthy individuals carrying O+ blood group. According to the general theories on the contribution of T lymphocytes in viral infection caused by influenza virus it is thought that lymphocytes: 1) are involved in the production and release of interferon, which protects other virussusceptible host cells [20]; 2) play the role of helper T cells for the differentiation of B-cells to synthesize antibodies against the HA antigen [21]; and 3) act as cytotoxic T cells involved in the killing of host cells expressing virus coded antigens on their cell surfaces [22] (i.e., cytolysis of infected cells [23].
An additional mechanism has been proposed for lymphocyte involvement in the limitation of and recovery from influenza virus infection. Lymphocytes have the ability to infiltrate tissues at susceptible sites or organs. If lymphocytes have specific receptors for the virus, but do not fully support virus replication, this may effectively reduce viral infectivity in the areas of highly susceptible cells [24,25]. The demonstration of influenza virus replication within peripheral blood lymphocytes may have important implications in understanding the outcome of influenza virus infection in vivo .
In order to evaluate any contribution of peripheral blood lymphocytes in pathogenesis of influenza virus, human peripheral blood mononuclear lymphocytes were cultured in the presence of PHA (mitogen), and exposed in vitro to influenza A subtypes H1N2, H5/H7-N1/N2, or H9N2 viruses. After the adsorption of the human lymphocytes with different influenza strains, the lymphocytes were thoroughly washed with PBS to wash out all the unabsorbed viral particles. Eventually, all three isolates produced CPE after 24 hours of incubation indicated the propagation of flu virus in human lymphocytes (Figure 1). To further confirm the results, the supernatants of the infected lymphocytes was harvested and inoculated in the MDCK cells (conventionally use for the propagation of the Flu virus). These MDCK cells also produced CPE after 24 h of incubation confirming the influenza virus propagation in the human lymphocytes. Afterwards the results were further confirmed by Reverse transcription and RT-PCR (Figure 2) of RNA isolated from both the infected human lymphocyte and MDCK cells. These results strongly suggest that each of the three influenza viruses were able to replicate in human lymphocytes (Figure 2) and indicated the existence of polyvalent receptors on human lymphocytes for various influenza viral subtypes. These finding are consistent with previous in vitro research showing that mouse T lymphocytes have α-2,6 sialic acid-linked receptors for a variety of influenza virus strains [7]. In addition, our haemagglutination assay and RT-PCR results confirmed that these influenza viral subtypes propagated in the human lymphocytes. Therefore, our findings strongly suggest that influenza virus can infect and replicate in human lymphocytes in vitro.
Our findings are consistent with previous reports that lethal H5N1 infection in mice stimulates a robust, virus-specific CD8 T cell response in the respiratory tract, but these CD8 T cells fail to control viral replication and undergo early contraction [13]. We hypothesize that Influenza virus stimulates the virus-specific CD8 T cells, but the virus infection destroys the CD8 T cells before they are unable to fully participate in the human immune response.
Since H1N2 was procured from a flu patient, this strain was used for further experiments to test the antiviral activity of a honey, ginger, and garlic (HGG) mixture in human lymphocytes. Previously, we found that HGG reduced influenza virus replication in MDCK monolayers (data not shown). Similar results were obtained in this report when the antiviral activity of HGG was determined in cultured human lymphocytes. CPE, haemagglutination assays, and RT-PCR all suggested that influenza replication in human lymphocytes was decreased by HGG. Interesting, comparison of the “uninfected” and “uninfected with 5% HGG” panels in (Figure 1), suggests that HGG also stimulates the proliferation of uninfected T lymphocytes. This supports previous reports that ginger [26] and honey [27,28] have volatile oils which stimulate T cell proliferation in cell culture.
One potential mechanism for the inhibitory effect of HGG on influenza virus involves the HA molecule of influenza virus. The HA protein is essential for viral entry into host cells [29]. The H1 and H2 subunit components of HA are linked through disulphide bonds and this is required for the attachment of influenza virus to the cell surface [30]. The effects of reducing/oxidizing agents that are present in honey, garlic and ginger may disrupt the disulphide bonds between H1 and H2 that are required for the functional integrity of HA. For example, garlic has sulfur-containing compounds such as alliin, allicin, diallyl sulfide, diallyl disulfide, diallyl trisulfide, ajoene, and S-allylcysteine [31] which may react with the thiol groups of disulfide bonds and cause conformational changes of the HA receptors, thus making the virus unable to interact with the host cell surface. In addition, honey [32] and ginger [33] contains hydrogen peroxide, which can act as a potent antimicrobial agent.
The present findings confirm the potential of a mixture of crude extracts of honey, ginger and garlic as an effective anti-influenza agent and provide a potential scientific basis for the therapeutic use of honey, ginger and garlic against influenza virus. The in vitro results reported here suggest that dietary uptake of honey, ginger and garlic may be beneficial against clinical influenza virus infection. Honey, garlic and ginger are readily available, inexpensive, and have long been used as “home remedies” to treat influenza viral infections. The results presented here may help justify the continuing use of honey, garlic, and ginger in traditional medicines in different ethnic communities worldwide and the use of honey in modern medications such as cough syrups [34,35].
This study was supported by a grant from Higher Education Commision, Pakistan. This work was supported by Dr. Adeebul Hasan Rizvi and Dr. Rana Muzaffar for letting us to perform real-time experiments in Molecular Diagnostics and Immunology Laboratory at Sindh Institute of Urology and Transplantation, Karachi, Pakistan.