Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Review Article - (2013) Volume 1, Issue 1
Keywords: Osteoblastogenesis; Osteopetrosis; Calcineurin; Osteoblasts
Osteoclasts are the multinucleated giant cells that decalcify the bone matrix and degrade collagens by performing acid decalcification and proteolytic degradation, while osteoblasts produce bone matrix protein and maintain mineralization to counterbalance the action of osteoclasts [1,2]. Bone homeostasis is such a micro-sculpting process regulated by the intricate interplay between these two opposing cell types that are derived from the different lineage: osteoclasts from myeloid precursors of hematopoietic stem cell, and osteoblasts from the multipotent mesenchymal progenitors of bone marrow [1,2] (Figure 1). M-CSF (macrophage colony stimulating factor) maintains the survival of osteoclasts, while RANKL induces osteoclastogenesis. Osteoblasts produce both M-CSF and RANKL to regulate osteoclasts. A single base pair insertion of M-CSF receptor gene Csfm in mice is the cause of the recessive mutation osteopetrosis (op) [3]. In human, an activating mutation of rank gene and a deactivating mutation of opg (encoded for osteoprotegerin, OPG) gene cause familiar expansile osteolysis and Paget’s disease of bones [4]. OPG is a decoy receptor for RANKL that is also secreted by osteoblasts and inhibits the binding of RANKL to RANK, blocking the differentiation of osteoclasts [5]. Denosumab, a humanized antibody against RANK that blocks the binding of RANKL to RANK, decreases the osteoclastic activity and bone resorption, was approved for reducing skeletal-related events in patients with metastatic cancer [6-8]. For osteoblastogenesis, RUNX2 (runt-related transcription factor 2, also known as Cbfa1, Osf2 and AML3) is a core binding factor and the essential regulator that guides the chondrocyte maturation and the differentiation of osteoblasts [9-11]. RUNX2 requires Cbfβ for sufficient DNA binding and its haploinsufficiency causes human cleidocranial dysplasia [12,13]. Mouse with cbfβ deletion developed delay in chondrocyte maturation and endochondral and intramembranous ossification [12,13]. Twist, a class basic helix-loophelix protein that is involved in the progression and invasiveness of several cancers, serves as a negative regulator of RUNX2 by direct interaction to block its activity at the transcription level [14]. Deletion of Twist rescued some of the skeletal defects of RUNX2-null mice [14]. Osterix, a Zinc-finger transcription factor, is specifically expressed in the immature chondro/osteoprogenitor cells and mature osteoblasts, essential for both intramembranous and endochondral ossification, acts downstream of RUNX2 to promote osteoblastogenesis [15,16]. Figure 1 illustrates the regulatory interplay among these key molecule networks in the regulation of osteoclasts and osteoblasts.
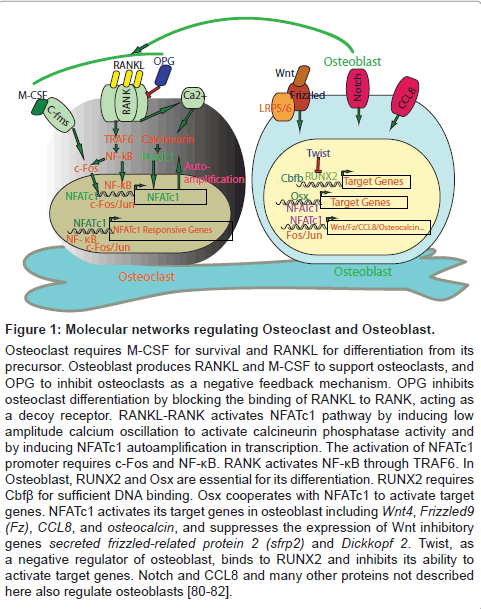
Figure 1: Molecular networks regulating Osteoclast and Osteoblast.
Osteoclast requires M-CSF for survival and RANKL for differentiation from its precursor. Osteoblast produces RANKL and M-CSF to support osteoclasts, and OPG to inhibit osteoclasts as a negative feedback mechanism. OPG inhibits osteoclast differentiation by blocking the binding of RANKL to RANK, acting as a decoy receptor. RANKL-RANK activates NFATc1 pathway by inducing low amplitude calcium oscillation to activate calcineurin phosphatase activity and by inducing NFATc1 autoamplification in transcription. The activation of NFATc1 promoter requires c-Fos and NF-κB. RANK activates NF-κB through TRAF6. In Osteoblast, RUNX2 and Osx are essential for its differentiation. RUNX2 requires Cbfβ for sufficient DNA binding. Osx cooperates with NFATc1 to activate target genes. NFATc1 activates its target genes in osteoblast including Wnt4, Frizzled9 (Fz), CCL8, and osteocalcin, and suppresses the expression of Wnt inhibitory genes secreted frizzled-related protein 2 (sfrp2) and Dickkopf 2. Twist, as a negative regulator of osteoblast, binds to RUNX2 and inhibits its ability to activate target genes. Notch and CCL8 and many other proteins not described here also regulate osteoblasts [80-82].
NFAT (Nuclear Factor of Activated T-cell) gene family was first identified in 1989 as a transcriptional factor activating interleukin-2 gene expression in response to T cell activation [17-19]. It contains four classic members: NFATc1 (also known as NFATc and NFAT2), NFATc2 (also known as NFATp and NFAT1), NFATc3 (also known as NFAT4), and NFATc4 (also known as NFAT3). The NFAT5 is structurally related but functionally distinct from the classic NFAT genes in which it does not require transcription partners and responds to osmotic stress as its main function [20,21]. The classic NFAT genes are uniquely expressed only in vertebrates. However, calcineurin, the activator of NFAT pathway, is expressed in non-vertebrates [17]. Calcineurin functional complex contains two subunits: the catalytic subunit A and the regulatory subunit B. Calcineurin-NFAT pathway plays essential roles in the regulation of immune system through modulating T cell response to stimuli and by regulating the development and function of T and B cells a well as other hematopoietic cells [18]. It also regulates numerous other biological systems by responding to sustained calcium influx from outside of the membrane induced by the activation of surface receptors such as RANK, receptor tyrosine kinases (RTKs, e.g T cell receptor, TCR), and G-protein coupled receptors (GPCR), etc. [18,19]. For example, NFAT signaling regulates heart development, skeletal and smooth muscle function, axonal migration, angiogenesis, renal function, metablosim, etc. [17-19]. NFAT proteins are heavily phosphorylated on the serine residues in the SRR and SP regions and are confined in the cytoplasm in the resting state. Activation of the surface receptors leads to the activation of Phosphoinositide Phospholipase C (PLC) that catalyzes the generation of inositol-1,4,5-trisphosphate (IP3) which interacts with its receptor (IP3R) on the surface of the endoplasmic reticulum (ER), inducing efflux of calcium from the ER and the subsequent depletion of the store calcium. This store calcium depletion activates the calcium sensor protein STIM1 or STIM2 on the ER [22-25], which then form oligomers and move to the ER and the plasma membrane junction, interact with the CRAC (calcium-released calcium channel) channel Orai, hence, inducing a sustained influx of calcium to activate calcineurin-NFAT signaling [26,27]. Deletion of STIM impairs calcium homeostasis and NFAT activation [28]. How RANKL-RANK signaling activates calcium oscillation is poorly understood, a recent study identified Tmem64 as a positive regulator [29]. Tmem64 is a transmembrane protein and was shown to interact with sarcoplasmic endoplasmic reticulum Calcium ATPase 2 (SERCA2), and its deficiency caused impaired RANKL-stimulated calcium oscillation [29]. It is currently not clear if RANKL-RANK signals through STIM and Orai for calcium oscillation. It is likely other molecules are involved in this important pathway. The calcium/calmodulin/calcineurin complex dephosphorylates NFAT proteins in the cytoplasm, rapidly propels the dephosphorylated NFAT proteins into the nucleus within minutes to exert their transcriptional action [30,31]. Cyclosporine and FK506 inhibit this nuclear translocation by forming a complex with cyclophilin A or FK506 binding protein (FKBP12) respectively that competitively binds to calcineurin and inhibits its phosphotase activity [30]. In response to diminishing receptor occupancy on the membrane and hence decreased calcium influx, NFAT proteins are rephosphorylated by several kinases including GSK3, PKA, MEKK, JNK, and others upon the termination of surface signaling and are exported back to and confined in the cytoplasm in an inactive state [19,31]. Figure 2 illustrates the NFAT signaling pathway activated by calcium-calcineurin and how RNAKL-RANK signaling may integrate into this pathway. The NFAT’s weak DNA domain (Rel Homology Domain, RHD) requires nuclear partners for transcription, rendering it a highly versatile functional regulator.
Figure 2: RANKL-RANK signaling and the integration of calciumcalcineurin- NFAT pathway.
Trimeric RANKL molecules bind to RANK, stimulate trimerization of RANK and its downstream signaling including activation of calcium, and subsequent calcineurin-NFAT signaling. Tmem64 positively regulates the RANKL-RANK activation of calcium influx, but how this occurs and if it signals through STIM and Orai remain unclear. RANKL-RANK activates NFκB and c-Fos, propelling their nuclear translocation, which cooperatively activates expression of NFATc1. NFATc1 is also capable of auto-activation with the aid of c-Fos (NFATn), forming a positive feedback loop. Calcineurin is inhibited by cyclosporine A (CsA) and FK506 and by DSCR1 (Down Syndrome Critical Region 1). Dephosphoorylaton of NFAT protein leads to rapid nuclear translocation and with its nuclear partners (NFATn), activates gene expression. Rephosphorylation of NFAT protein by GSK3, PKA, DYRK, CK1, and others promote nuclear export and termination of NFAT signaling. When the negative inhibition by GSK3 (ec. changing the residues of the GSK3 phosphorylation sites) is removed the positive NFATc1 feedback loop becomes unopposed and the NFATc1 signaling is rendered constitutive. Enhanced NFATc1 activity in osteoclasts leads to osteopenia while in osteoblasts causes osteopetrosis [19,39,65].
It is now clear that calcium-calcineurin-NFAT pathway plays critical roles in bone health by maintaining balanced regulation of osteoclastic and osteoblastic activities. RANKL-RANK signaling stimulates NFATc1 to induce the expression of a number of genes within osteoclasts to activate differentiation of osteoclast leading to bone resorption and remodeling. These findings explain many of the clinical observations with regard to the effect of cyclosporine, the inhibitor of calcineurin-NFAT pathway.
NFATc1 as the master transcription regulator of osteoclasts in response to RANKL-RANK signaling
In the early 1990’s, by studying the c-fos knockout mice it was unexpectedly found that these mice lost the ability for osteoclast differentiation in the bone marrow and developed osteopetrosis, transplantation of normal bone marrow or ectopic expression of c-fos rescued the defect [32,33], however, the mechanism of this phenotype remained unclear until the important discovery of NFATc1 as the master transcription regulator of osteoclasts in 2002 through gene profiling of RANKL-induced osteoclast differentiation [34]. Expression of NFATc1 rescued osteoclast precursors lacking c-fos, while NFATc1-deficient fetal liver or embryonic stem cells were not capable of rescuing osteoclastogenesis of the c-fos-deficient mice [35]. NFATc1 is induced and activated by RANKL during osteoclastogenesis. RANKL is a cytokine of TNF superfamily, expressed by osteoblasts as type II membrane-associated protein ligand and is recognized by its receptor RANK in the osteoclast precursors during osteoclast maturation. RANKL induces a sustained low amplitude calcium oscillation to activate calcineurin-NFATc1, promoting the nuclear import of NFATc1 to form an autoregulatory feedback system for enhancing its own expression as well as activating a battery of other genes essential for osteoclastogenesis, aided by c-fos [34,36]. The transcriptional activation of NFATc1 requires TRAF6, NF-κB, and c-fos in osteoclasts. RANKL induces TRAF6 to activate NF-κB pathway which in turn activates the expression of NFATc1 whose promoter bears an NF-κB binding element [34]. Thus, RANKL utilizes two pathways to activate NFATc1 activity, one through calcium-calcineurin, and another through TRAF6-NFκB. TRAF6-deleted mice develop osteopetrosis consistent with its role in activating NFATc1 transcription in osteoclasts [37]. An inhibitor of NF-κB down-regulates NFATc1 expression to inhibit osteoclastogenesis [38]. NFATc1 is the convergent point of RANKL-RANK-stimulated signaling pathways for inducing osteoclastogenesis (Figure 1).
Deletion of NFATc1 gene from osteoclasts reveals gene transcription programs regulated by NFATc1
The Nfatc1-knockout mice die of heart valve defect, when rescued by Tie-2-promoter-driven expression of NFATc1 (NFATc1-/-;Tie2-NFATc1+) showed profound osteoclastogenesis defect with delayed bone formation [39]. Adult mice with conditional deletion of NFATc1 in osteoclasts developed osteopetrosis due to the deficiency in osteoclastogenesis, in part owing to the de-repression of RANKL negative regulator OPG that was normally suppressed by NFATc1 [40]. Osteoclast NFATc1 regulated two sets of transcription program, one dependent on NFATc1, including genes such as Oscar, Calcr, Itgb3, Rcan2, Myo1d, and Mst1r, and another independent of NFATc1, but augmented by it including genes like Mmp9, Acp5, Ctsk, Mmmp14, and Clcn7 [40]. All these results demonstrate the critical importance of NFATc1 as the master transcriptional regulator of osteoclasts. How the two programs synchronize to orchestrate the differentiation of osteoclasts in both temporal and spatial fashion will be an interesting subject for further investigation. Specific deletion of each of the component gene may offer further insight into how much each component gene of the programs contributes to the differentiation process and what segment of function they exert. Recently, analysis of IP3R2-deleted mice showed osteoblasts in a co-culture system were capable of inducing NFATc1 activation in osteoclasts independent of calcium oscillation [41]. This interesting observation suggests a secondary mechanism for activating NFATc1 in osteoclasts that warrants further investigation.
Regulation of NFATc1 by other molecules in osteoclasts
The role of NFATc1 in osteoclastogenesis is counteracted by many other molecules. RANKL inhibits the expression of Id1, Id2, and Id3 [42]. Over expression of all these three genes in the bone marrow-derived monocyte/macrophage lineage cells reduces the formation of osteoclasts [42]. Overexpression of Id2 decreased the stimulation of NFATc1 by RANKL [42]. Similarly, RANKL inhibited expression of MafB during osteoclastogenesis and overexpression of MafB resulting in reduced DNA binding activity of c-fos and NFATc1 and hence the activation of NFATc1 and other genes such as OSCAR (osteoclast-associated receptor) [43]. IRF-8 (interferon regulatory factor-8) blocked NFATc1-induced osteoclastogenesis and was suppressed by RANKL, its deletion caused severe osteoporosis in mice [44]. Bcl-6 inhibited osteoclastogenesis by suppressing the expression of NFATc1 and other genes, its deletion caused osteoporosis in mice [45]. Consistent with this, Blimp1 (B lymphocyte-induced maturation protein-1) inhibited BCL-6 in osteoclasts, and its deletion caused osteopetrosis [45]. The Blimp1-Bcl-6 axis appears to be dependent on calibrating NFATc1 expression for regulating bone homeostasis. The heterozygous mice with an R740S mutation in the a3 subunit of V-ATPase showed mild osteopetrosis, owing to increased lysosomal pH in osteoclasts which impairs nuclear translocation of NFATc1 by increasing the expression of RCAN, an inhibitor of NFATc1 [46]. This study highlights the importance of NFATc1 nuclear translocation during osteoclast maturation and that the factor impacting NFATc1 activity resides as far as lysosome. All these experimental evidence demonstrate that NFATc1 is the central converging point for osteoclastogenic regulation by multiple pathways.
Role of NFATc1 in cherubism
The genetic model cherubism has also provided important insight into the regulation of bone homeostasis by NFATc1 in osteoclasts. An autosomal dominant pediatric disease that is characterized by increased symmetrical bone resorption of the maxilla and mandible with fibrous tissue deposit that causes cystic changes of the bones, cherubism is caused by a mutation of SH3-doman binding protein 2 (Sh3bp2) in 80% of inflicted patients [47,48]. Cherubism mice display generalized osteoporosis with extensive inflammation related to an elevated TNFα level and an increased myeloid response to M-CSF and RANKL, and consequently increased activation of NFATc1 [49]. The mutation of Sh3bp2 renders itself resistant to the tankyrase-mediated ubiquitination and hence results in its enhanced stabilization leading to the subsequent hyperactivation of the SRC, SYK, and VAV signaling pathways [49,50]. When NFATc1 was deleted from the bone marrow cells driven by the Mx1-CRE, the osteoporosis phenotype of the cherubism mice was reversed, owing to the suppression of osteoclastogenesis in the absence of NFATc1, while there was no impact on the inflammation that was dependent on TNFα [50]. There is evidence that TNFα also activates the NFATc1 pathway in macrophage [51]. How this may be related to bone remodeling during inflammation requires further studies. DRYK1 is a dual specificity kinase that inhibits NFATc1 in both osteoclasts and osteoblasts, while NFATc1 induces expression of DYRK1 as a negative feedback mechanism. DYRK1 transgenic mice exhibited reduced osteoclastogenesis but still developed osteoporosis due to the overriding impact of defective osteoblastogenesis [52]. The expression of DYRK1 is increased by 1.5 fold in the Down Syndrome patients who commonly have bone loss [53].
Regulation of NFATc1 and osteoclasts by inflammation
Chronic inflammation is frequently associated with bone destruction and osteoporosis, one mechanism of which is related to the increased serum level of several cytokines including TNFα [54]. While TRAF6 is essential in mediating the activation of NFATc1 by RANKL, TRAF2 appears essential for TNFα-stimulated osteoclastogenesis. The deficiency of TRAF2 in mice also mildly impaired osteoclastogenesis [55]. A recent study found that macrophage inflammatory protein MIP-1δ was involved in mediating pathological bone destruction of renal cell carcinoma bone metastasis and rheumatoid arthritis, by enhancing NFATc1 activation and osteoclastogenesis [56]. Ligands that activate TLRs are found to activate osteoclasts and enhance bone resorption [50]. In certain conditions, TLRs and certain cytokines inhibited RANK, RANKL and osteoclasts. For example, IL-10, a immune modulator, inhibited osteoclasts by preventing nuclear translocation of NFATc1 and RANKL-induced expression of NFATc1, Jun and Fos [57], IL4 and IL27 shared the similar mechanism in suppressing NFATc1 induced by RANKL [58,59], and TLRs and γ-interferon inhibited RANK and osteoclast precursors [60]. In addition, in osteoclasts, it has been shown that Toll-like receptor 2 (TLR2) that mediates inflammation and bone metabolism by activating RANKL, NFATc1, NF-κB, and others [61]; protein inhibitor of activated STAT3 (PIAS3) inhibits the promoter of NFATc1 [62]; Notch recruits NF-κB p65 to the promoter of NFATc1 for transcription activation [63]; and vitamin D inhibits the NFAT transcriptional activation [64].
It may appear paradoxical for NFATc1 to play positive roles in both osteoclast and osteoblast, two opposing cell types essential for bone homeostasis. The role of NFAT pathway in osteoblastogenesis appears more complex than in osteoclastogenesis. While NFATc1 is essential and sufficient for osteoclastogenesis, functioning as a master transcription regulator that integrates several signaling pathways, it appears to be one of the many pathways controlling osteoblast maturation.
NFATc1 signaling, Wnt pathway and chemokine expression in osteoblasts
A mutant mouse line (NFATc1nuc transgenic mice) that carried a non-exportable constitutively nuclear NFATc1 in osteoblasts developed osteopetrosis, with severely increased bone mass associated with massive osteoblast proliferation and overgrowth [39,65]. The enhanced proliferation of osteoblasts increased production of many monocyte chemoattractants and stimulated a coordinated reprogramming of the gene expression that controls the Wnt signaling pathway. Despite NFATc1nuc was not expressed in osteoclasts, there was increased osteoclastogenesis with normal expression of RANKL and OPG. Hence, both bone formation and bone absorption were increased in the mutant mice, with bone formation as the overriding predominant process. The critical regulator of osteoblast RUNX2 showed no change in expression. The expression of the chemoattractants CCL8, CCL6, and CCL12 was increased, CCL8 being a direct target of NFATc1 [39]. The Wnt/Wingless signaling pathway was induced coordinately, characterized by increased the expression of Wnt4 and Frizzled9 and decreased the expression of the Wnt inhibitors secreted frizzled-related protein 2 (sfrp2) and Dickkopf 2 (DKK2) [39]. The Wnt proteins signal through their transmembrane co-receptors encoded by the low-density lipoprotein receptor-related protein (LRP5/6) and Frizzled (FZD) gene families. A constitutively active Lrp5 gene construct was previously shown to promote osteoblast differentiation independent of RUNX2 pathway [66]. The mice deficient of LRP5 showed decreased osteoblast differentiation and osteopenia [67]. The mutation of Lrp5 is the cause of the human autosomal recessive disorder osteoporosis-pseudoglioma syndrome (OPPG) [68]. Inhibition of Wnt signaling impairs osteoblastogenesis and bone formation [69]. Strontium ranelate (SrRan), a drug that increases bone formation and inhibits bone resorption, used clinically to palliate bone pain caused by metastatic cancer such as prostate or breast cancer, was recently shown to induce NFATc1 nuclear translocation and increase the expression of Wnt3a, Wnt5a, and β-catenin in osteoblasts [69], further linking these two pathways into the signaling network of bone homeostasis regulation. However, the exact interaction between these two major pathways in bone homeostasis is far from being clear, further studies using complementation experiments may determine if NFATc1 is sufficient to rescue the Wnt signaling defect in bone homeostasis and if Wnt signaling components are essential for the function of NFATc1 in osteoblasts (Figure 1).
Role of calcineurin in osteoblast regulation
Calcineurin consists of catalytic subunit A and regulatory subunit B, both of which are critical for its function [18,19]. Consistent with the stimulatory effect of NFATc1 in osteoblasts, calcineurin subunit Aα, one of the calcineurin catalytic subunit, has been shown to stimulate osteoblast in calvarial culture system when overexpressed in vitro [70]. Mice with calcineurin Aα deleted developed severe osteoporosis associated with decreased expression of RUNX2, osteoclacin and bone sialoprotein, and the expression of these proteins were also suppressed by FK506 treatment [70]. Osteoblast expresses all the calcineurin A and B isoforms [70]. It is not clear if the other two isoforms of calcineurin A (β and γ) have similar effect on osteoblasts. It is surprising to see the severity of osteoporosis with only when the α isoform was deleted. Separately, when calcineurin B1 (CnB1) was deleted specifically from osteoblasts, mice developed osteopetrosis phenotype with an increased OPG production, decreased RANKL, and decreased osteoclastic activity [71]. The CnB1-deficient osteoblasts showed inhibition of NFATc1 dephosphorylation, hence decreased nuclear translocation [71]. Deletion of the calcineurin inhibitor RCAN2 caused osteoblast defects in mice with delayed intramembranous ossification and impaired cortical bone formation, similar to the mice lacking the type 2 deiodinase thyroid hormone activating enzyme or with dominant-negative mutations of thyroid hormone receptor α(TRα) [72]. These results suggest that the involvement of calcineurin-NFAT pathway is likely much more complex in osteoblasts with different isoform and possibly different NFAT protein exerting distinct function. In a separate study by the same lab, it was found that NFATc1 suppressed the promoter of osteocalcin during osteoblast differentiation possibly through a HDAC mediated mechanism [73]. Histone deacetylase (HDAC) 3 has been shown to interact with the amino terminus of Runx2 to repress osteocalcin promoter and osteoblast maturation [74]. Mice with HDAC3 deleted by Osx-Cre developed decreased bone mass and osteoblasts but increased adipocytes in the bone marrow [75]. How epigenetics and chromatin remodeling regulate bone homeostasis remains poorly understood and shall be a major area for future studies.
Cooperation between Osterix and NFATc1 in osteoblastogenesis
As illustrated in figure 1, the essential osteoblast transcription factor Osterix cooperates with NFATc1 to regulate osteoblastogenesis by binding to the promoters of their target genes to form a cooperative transcription complex [76]. Osx acts downstream of RUNX2, and like RUNX2, Osx-null mice lack bone formation with absence of osteoblastogenesis while preserving functional osteoclasts [77]. Overexpresssion of NFATc1 in osteoblasts stimulated the transcription of Osterix-dependent genes such as type I collagen, exposing osteoblasts to FK506 reduced the bone formation of the mice [76]. Though FK506 inhibited both osteoclast and osteoblast, the inhibition on osteoblasts was predominant [76]. This explains at least in part the clinical observation that patients receiving immunosuppressants after organ or bone marrow transplantation frequently developed osteoporosis [78].
Regulation of NFATc1 and osteoblasts by reproductive hormones
It is well established that estrogen and testosterone have major impact in bone health. Recently, it was found that the skeletal system regulated the production of testosterone in males (but not estrogen in the ovaries) through osteoblasts-secreted osteocalcin that bond to its GPCR and activated testosterone synthesis through a CREB-dependent manner in Leydig cells [78]. This is fascinating and demonstrates that the skeletal system and the endocrine system reciprocally regulate each other. Since NFATc1 regulates the promoter of osteocalcin [70,73,79], one may suspect whether or not NFATc1 may also play an indirect role in regulating testosterone level and impacting bone health in males.
Notch signaling regulates NFATc1 and NFATc2 in osteoblasts
Notch signaling also plays important role in bone homeostasis through regulating NFAT as we as other pathways. Notch signaling inhibits osteoblasts, and the deletion of transcription factor RBPjk, a mediator of all canonical Notch signaling, and the deletion of Hey1 and HeyL, two target genes of RBPjk, caused high bone formation [80]. Hey1 bond to the promoter of NFATc1 and suppressed its expression. Expression of NFATc1 was increased in the bone of RBPjk deleted mice [81]. These results indicate that Notch signaling suppresses NFATc1 in osteoblasts to prevent excessive bone formation. Interestingly, NFATc1 inhibits Notch signaling in osteoblasts. Therefore these two signaling pathway reciprocally inhibit each other to regulate the balance of bone formation by controlling the activity of osteoblasts. NFATc2 also appears to inhibit Notch signaling, and Notch inhibits NFATc2 likely as a negative feedback loop in osteoblasts [82]. These results indicate a complex regulatory network between NFAT and Notch and perhaps other signaling pathways in regulating the proliferation and differentiation of osteoblasts.
Roles of NFATc2 and NFATc3 in osteoblasts
NFATc2 may be involved in the regulation of osteoblasts as it was found to be a target gene of Nell-1. How exactly NFATc2 regulates osteoblasts is poorly understood and remains to be explored. NFATc2 acts to stimulate NFATc1 transcription upon RANKL activation in osteoclasts prior to the initiation of autoamplification of NFATc1 [35,36]. Nell-1 is a growth factor required for the bone formation and a target gene of RUNX2. Nell-1 stimulates expression of extracellular matrix proteins that are required for bone and cartilage cell differentiation [83]. NFATc3 may play a role in bone homeostasis through responding to high extracellular calcium level and stimulating RANKL production in osteoblasts which in turn drives the activity of osteoclasts [84]. The roles of NFATc2, c3 and c4 in bone homeostasis are not well understood and further studies are needed.
In summary, we have reviewed the roles of calcineurin-NFAT signaling pathway in bone homeostasis. NFATc1 is critical for both osteoclastogenesis and osteoblastogenesis. The roles of other NFAT molecules are less clear and require further investigation. Manipulating NFAT signaling through regulating molecules that regulate NFAT pathway (ec. GSK3, calcineurin, etc.) or through direct inhibition of NFAT proteins may provide avenues for intervening bone health. Further studies to understand the crosstalk between the calcineurin-NFAT pathway and the other major signaling pathways as well as chromatin remodeling factors during osteoclastogenesis and osteoblastogenesis shall provide further insight into the mechanism of bone formation and remodeling and the stem cell biology of the bones.