Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Original Research Article - (2023)Volume 11, Issue 3
Background: The global annual population of newborn with structural haemoglobin disorders is estimated at five million and Nigeria accounts for more than 30% of this with under-five mortality from haemoglobinopathies reaching 50%-90%. Despite this huge burden and a 15-fold reduction in deaths from haemoglobinopathies in countries that conduct newborn screening, most sub-Saharan Africa countries do not have a screening program. The widely used diagnostic haemoglobin electrophoresis is also not sensitive for newborn screening.
Objectives: This study was carried out to determine haemoglobin phenotype patterns and frequency in neonates attending routine immunization clinics in Bida, and to identify factors associated with the occurrence of haemoglobinopathy.
Materials and Methods: It was a descriptive cross-sectional study that recruited 254 neonates by multi-staged sampling technique from nine immunisation centres. Heel prick blood sample collected on Guthrie cards were tested using High-Performance Liquid Chromatography (HPLC). The relationship of various risk factors with the occurrence of an abnormal haemoglobin variant was analysed with the Statistical Package for Social Sciences.
Results: The Hb phenotypes found in this study were HbFA-73.6% (187/254), HbFAS-23.2% (59/254), HbFAC-1.6% (4/254), HbFS-1.2% (3/254), and HbFAD-0.4% (1/254). There was an almost equal abnormal haemoglobin occurrence in both genders. The majority (89%) of mothers did not know their Hb phenotype, 25% of these had a newborn with an abnormal phenotype and 20% were married in consanguineous marriages. Wrong perception of sickle cell disease was also common.
Conclusion: Abnormal haemoglobin variants were present in more than one-quarter (26.4%) of the neonatal population studied in Bida. Most parents were not aware of their haemoglobin phenotype and had a wrong perception of sickle cell disease. Consanguinity though common in the population did not significantly affect the occurrence of an abnormal haemoglobin phenotype.
Newborn screening; Sickle cell; Chromatography; Hb variant
Haemoglobin (Hb) is a protein molecule in red blood cells that functions in gaseous exchange and maintenance of the body buffer system [1,2]. Abnormalities of haemoglobin can be structural or quantitative i.e. structural variants or thalassemias [3]. The structural abnormalities are the most common monogenic disease with approximately 5% of the world population being carriers [4]. The Sickle Haemoglobin (HbS), Haemoglobin-C (HbC), and Haemoglobin-E (Hb E) are the commonest structural variants that have a significant health impact worldwide [5,6]. Sickle Cell Disease (SCD) is a group of inherited red blood cell disorders caused by the inheritance of two abnormal haemoglobin genes, one of which is HB S. It results in the production of abnormal (sickle) shaped red blood cells and chronic haemolytic anaemia [1] The common SCD phenotypes include Hb SS, Hb SC, and Hb Sßthalassemia, the less commonly occurring ones are Hb SD, HbSE, and Hb SO. Sickle cell anaemia (HB SS) is the most severe form of sickle cell disease [3]. It occurs with the inheritance of two genes that code for haemoglobin S. The sickle cell trait (Hb AS) occurs with the inheritance of one copy of the sickle gene and one normal gene. It is not a type of sickle cell disease and is usually asymptomatic [1]. It may however produce sickle-shaped red blood cells in extreme physical or environmental conditions. Globally, about 5.7 million neonates are estimated to have structural haemoglobin variants in different combinations, and three to four hundred thousand babies are born with severe forms of these variants each year [7]. About 75%-85% of these come from Africa especially Nigeria where 150,000 children are born annually with haemoglobinopathy amounting to 33% of global annual births [8,9]. In Nigeria, 2%-3% of the total newborn population are sufferers and 24.7% are carriers [10,11].
Under-5 mortality from haemoglobinopathies and its complications reach 8%-16% [11] in Sub-Saharan Africa with death occurring before the fifth birthday in 50%-90% of undiagnosed children who suffer sickle cell anaemia [12,13]. The World Health Organization (WHO) has linked the reduction in morbidity and mortality from haemoglobinopathies to early diagnosis via Newborn Screening (NBS) combined with comprehensive medical care [14]. In Nigeria however, the average age at diagnosis of the most common Haemoglobinopathy (Hb SS) is 27.33 (± 26.36) months [9] and the most widely available method of haemoglobin genotype testing is electrophoresis using cellulose acetate paper. This method has low diagnostic sensitivity before six months of life, does not quantify haemoglobin, and is less reproducible. The High-Performance Liquid Chromatography (HPLC) and Isoelectric Focus (IEF) methods do not have these limitations [15]. This study aimed to determine the haemoglobin phenotypes, their frequency of occurrence, and the factors associated with the occurrence of an abnormal variant occurring in newborns attending Bida community immunization centres using the HPLC method.
It was an observational, cross-sectional study that screened 254 term neonates across nine immunization centres from six wards in Bida local government of Niger State (Figure 1).

Figure 1: Map of Bida local government area showing the
wards where immunization centres were selected. 
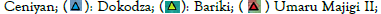

The study was registered with the Health Research Ethics Committee of the Federal Medical Centre Bida with protocol number FMCB/HCS/HREC/APPR/VOL1/5/18 and Niger State Ministry of Health research ethics committee in May 2018. The study period was from June 2018 to December 2019, the study participants were recruited over four months (September - December 2019).
The study sites and sample population were selected by a multistage sampling method from twenty-eight registered immunization centres with an average annual population of 10,020 vaccinated neonates (Figure 1). The minimum sample size of 254 was calculated using the standard formula for a single proportion [16].
Heel prick samples were collected on Guthrie cards from term neonates on routine immunization visits that had not been previously transfused with blood or red blood cells. The samples were analysed with the VARIANTnbs Newborn Screening machine at the HPLC North-central Nigeria regional laboratory, Keffi, Nasarawa state [17]. A structured questionnaire was used to collect data on sociodemography, parents’ knowledge and phenotype as well as other factors associated with abnormal variant occurrence. These were analysed with the Statistical Package for the Social Sciences (SPSS) version 23.0. Frequencies, proportions, and percentages were computed for categorical variables of interest like haemoglobin phenotype pattern and parental knowledge of haemoglobin phenotype. The prevalence of the various haemoglobin patterns was expressed in charts and tables. Chi-square (χ²) test was used to determine associations between the outcome variable of interest (haemoglobin phenotype) and other categorical independent variables of interest at a 5% level of significance (p-value <0.05). Fisher's exact test was used in place of Chi-square if the expected frequency was <5.
The median age at diagnosis was 7 days (IQR 7), with a male to female ratio of 1.04:1. The various Haemoglobin (Hb) patterns found in this study include Hb F (Foetal Haemoglobin), Hb A, Hb S, Hb C, and Hb D combined in various homozygous and heterozygous forms as shown in (Figure 2).

Figure 2: Frequency of identified haemoglobin phenotypes.
Normal haemoglobin FA was the major phenotype 73.6% (n=187), and the carrier phenotype Hb FAS 23.2% (n=59) was the most common structural variant found in the study population. Hb FS, which is the homozygous sickle cell anaemia and the most severe sickle variant, occurred in 1.2% (n=3) of the population. Haemoglobin AC and AD were the non-sickled phenotype variant observed. The study showed that 26.4% (n=67) of the study population had an abnormal haemoglobin variant which included (59 Hb FAS-23.2%, 3 Hb FS-1.2%, 4 Hb FAC-1.6%, 1 Hb FAD-0.4%)
The combined prevalence of sickle haemoglobin (FAS, FS) in the study population was 24.4% (n=62) and the sickle carrier phenotype was the predominant variant (Table 1).
| Parameter | Frequency affected N=67 | % abnormal variant in affected subjects | % abnormal variant in total study population N=254 |
|---|---|---|---|
| Abnormal hemoglobin pattern | |||
| Sickle hemoglobin | |||
| FAS | 59 | 88 | 23.2 |
| FS | 3 | 4.5 | 1.2 |
| Total sickled hemoglobin | 62 | 92.5 | 24.4 |
| Non-Sickle hemoglobin | |||
| FAC | 4 | 6 | 1.6 |
| FAD | 1 | 1.5 | 0.4 |
| Total non-sickled hemoglobin | 5 | 7.5 | 2 |
| 100 | 26.4 | ||
Table 1: Frequency of abnormal variants in the affected population.
A majority 80.3% (n=209) of mothers were aware of the term ‘Sickler’ (Figure 3 and Table 2), but only a few 15.4% (39/254) had the correct knowledge that it referred to persons with a blood disorder.

Figure 3: Awareness of the word ‘Sickler’ and its meaning.
| Parameter | Normal hemoglobin N (%) | Abnormal hemoglobin N (%) | χ² | p-value |
|---|---|---|---|---|
| Awareness of the word ‘sickler’ | ||||
| Yes | 151(59.4%) | 52(20.5) | ||
| No | 36(14.2%) | 15(5.9%) | 0.302* | 0.596 |
| Meaning of sickler | ||||
| Don’t know | 55(21.7%) | 21(8.3%) | ||
| Skinny person | 18(7.1%) | 11(4.3%) | ||
| Falls sick very often | 84(33.1%) | 26(10.2%) | ||
| A person with a blood disorder | 30(11.8%) | 9(3.5%) | 2.699* | 0.44 |
Note: (*) Pearson’s chi-square
Table 2: Relationship between knowledge of haemoglobinopathies and abnormal phenotype.
Abnormal haemoglobin variant was found in sampled neonates from all the ethnic groups. Those born to parents of Nupe ethnicity accounted for 22% (n=56) of neonates with an abnormal variant in the total sampled population. The relationship between ethnicity and an abnormal variant was however not statistically significant. Ten percent (10%) (n=26) of the neonates with an abnormal haemoglobin variant were delivered to multiparous women who had between two and four children, a higher birth however did not show a further rise in the occurrence of an abnormal variant (Table 3). Consanguineous marriage was a common practice as 20% of caregivers were married in this setting but its effect on the occurrence of an abnormal variant was not significant.
| Parameter | Normal hemoglobin N=187 n(%) | Abnormal hemoglobin N= 67 n(%) | χ² | p-value |
|---|---|---|---|---|
| Ethnicity | ||||
| Nupe | 163(64%) | 56(22%) | ||
| Gwari | 3(1.2%) | 1(0.4%) | ||
| Hausa | 7(2.8%) | 5(2.0%) | ||
| Yoruba | 9(3.6%) | 2(0.8%) | ||
| Igbo | 3(1.2%) | 2(0.8%) | ||
| Others (Kambari) | 2(0.8%) | 1(0.4%) | 3.160** | 0.689 |
| Social class | ||||
| High | 48(19%) | 20(8.0%) | ||
| Middle | 88(35%) | 27(10%) | ||
| Low | 51(20%) | 20(80%) | 1.704* | 0.636 |
| Parity | ||||
| One | 48(19.0%) | 17(6.7%) | ||
| Two-four | 96(38%) | 26(10%) | ||
| Five-seven | 28(11%) | 16(6.3%) | ||
| Greater than 7 | 15(5.9%) | 8(3.1%) | 4.710* | 0.194 |
| Consanguinity | ||||
| Yes | 41(16%) | 9(4.0%) | ||
| No | 146(57%) | 58(23%) | 2.250* | 0.134 |
Note: (*) Pearson’s chi-square; (**) Fisher’s exact test
Table 3: Factors associated with the occurrence of haemoglobinopathy.
This study of haemoglobin phenotype patterns among neonates on their first immunization visit in Bida, Niger state revealed the presence of normal (HbFA) as well as abnormal haemoglobin variants in homozygous and heterozygous combinations. These abnormal variants were Hb FAS, Hb FAC, Hb FS, and Hb FAD in order of decreasing frequency. Our observations were comparable to a community-based study by Inusa BP, et al. [10] that used the HPLC method to screen newborn in Northern Nigeria and Adeyemo T, et al. [18] in Lagos, Nigeria that reported abnormal phenotypes SS, SC, SD Punjab, among older sickle cell disease patients aged 2-15 years using HPLC method as well. Both studies reported the Hb Sβ-thalassemia which was not found in our study that focused on structural variants. The study by Odunvbun ME, et al. [19] in Benin city South-South Nigeria also reported similar pattern using isoelectric focusing method, although there was no occurrence of the Hb D variant. Nnodu OE, et al. [15] reported Hb A, S and C in different combinations in their community-based study in North-Central Nigeria but the ELISA kits used in their study are designed to detect these three Hb types only. These reports may imply that the haemoglobin pattern in different parts of Nigeria is similar with slight variation in some geo-political zones. This similarity would make it easy to design strategies for neonatal screening campaigns that would be employable nationwide.
The frequency of normal HBAA found in our study was comparable to the previous reports from different parts of Nigeria where newborn screening had been carried out using either the HPLC or IEF methods [10,19,20] therefore, a significant majority of newborns in Nigeria have normal haemoglobin despite the occurrence of abnormal haemoglobin variants. The combined prevalence of abnormal haemoglobin variants S, C, and D in this study was 26.4%. The obtained sickle cell disease prevalence and the national prevalence of sickle cell disease were also comparable (1.2% vs. 2%) [22]. An earlier report from Northern Nigeria by Fleming AF, et al. [21] in 1970, and a more recent report of abnormal haemoglobin variants from the same region by Inusa BP, et al. [10] reported a 26.7% and 24.2% prevalence respectively, which were comparable with our findings. Abnormal haemoglobin prevalence has remained high in the country despite the recommendations on the need to increase awareness and encourage premarital screening.
The average annual newborn population in Bida was 10,000 births [23] a sickle cell disease prevalence rate of 12/1000 (1.2%) live births amounts to 120 affected babies delivered per year. This places the community in a category that requires universal newborn screening. A report by Tshilolo L, et al. [23] stated that universal screening should be carried out in a population where there are 5 or more cases of sickle cell disease per 10,000 (0.5/1000) births. The prevalence of sickle cell disease in this study is 2.4 folds higher than the recommended threshold for universal screening [24] therefore concerted efforts in expanding facility for screening and comprehensive care are required to meet this enormous healthcare need.
The Sickle cell disease prevalence we reported was four times higher than that of a newborn screening study in South-Eastern Nigeria (1.2% vs. 0.3%) [20] and another from South-South Nigeria reported a prevalence that was 2.5 times higher than our findings(1.2% vs. 3.0%). The study methodology (HPLC vs. IEF/ Universal vs. targeted screening), different study settings (hospital vs. community-based screening), a low level of awareness of the availability of screening facilities, and an active campaign against at-risk marriages by religious bodies may have accounted for these observed differences. Studies in Africa have reported a sickle cell disease prevalence range of 1.2% to 1.5% [25] and 1%-1.1% in Southern America [26] but a much lower prevalence in Europe and other parts of America (0.01%-0.62%) [26]. The sickle cell gene is endemic where stable malaria transmission occurs hence the higher prevalence in sub-Saharan Africa where our study was carried out and South America. The impact of migration may account for the prevalence in Europe and other parts of America as most of the babies with the disorder in these regions were born to immigrant women of Sub- Saharan, Asian and Hispanic descent [26].
The most significant reported risk factor for developing sickle cell disease in the newborn is the occurrence of the carrier state in both parents as the disease is inherited in an autosomal recessive pattern. There is a 25% chance of an offspring developing sickle cell disease per pregnancy when both parents are heterozygous Hb S carriers [14]. Most parents (89%) in this study did not know their haemoglobin phenotype and this trend could increase the birthing of an affected offspring when sickle gene carriers who are unaware of their phenotypes get married. The parents of all the neonates who had sickle cell disease in this study did not know their haemoglobin phenotype and about one-third of the mothers who were unaware of their phenotype had babies with abnormal haemoglobin variants (HB C, D, S). This was comparable to a report by Odunvbun ME, et al. [27] where 63% of affected babies recruited in the study were born to mothers who were unaware of their Hb phenotype [19]. The study also reported low awareness of parental phenotype, the highest reported prevalence of sickle cell disease among neonates in Nigeria, as well as a high carrier state in the population studied. The level of awareness of parental phenotype in the study by Sam-Amoye [28] in Lagos, Nigeria was higher than the findings in this study and this may have accounted for the low carrier prevalence (14.8%) in the study. Sex chromosomes are not involved in the transmission of the sickle cell gene; gender is therefore not likely to be an associated factor for haemoglobinopathy as depicted by the almost equal male to female ratio. Newborn screening studies in other parts of Nigeria and Africa did not show any significant gender preponderance of abnormal haemoglobin variants.
The general lack of in-depth knowledge about what sickle cell disease is may have significant implications for its control. In this study, 80% of the mothers had heard of the word 'Sickler' before but more than half of these did not know it was an inherited blood disorder. This was similar to a report by Odunvbun ME, et al. [19]. This showed better awareness of sickle cell disease among educated mothers of under-five children. A large proportion of mothers in this study had no formal education as derived from their social class scoring; this may have influenced their knowledge of haemoglobinopathies as well as the need to do a premarital screening as a measure to prevent the risk of birthing an affected offspring. A higher level of formal education correlates with increased awareness of one's haemoglobin phenotype as reported by Piel FB, et al. [29] in their newborn screening study that showed a higher level of formal education among mothers and an associated increase in haemoglobinopathy awareness.
This study found that 20% of the parents of our sampled population were married in consanguineous settings with 4% (n=9) of the babies born to them inheriting an abnormal phenotype while 23% (n=58) of neonates born in nonconsanguineous settings had an abnormal phenotype. Consanguinity is widely considered to increase the gene frequency of any recessively inherited disorder in areas where the allele frequency is high [7,30,31] but this was not reflected in our findings and was comparable to the findings of a crosssectional study by El Mouzan MI, et al. [32] on the role of consanguinity in major genetic disorders in Saudi Arabia over 2 years. A possible reason suggested by the study on the lack of effect of parental consanguinity is that in areas of increased prevalence of SCD, and an even higher prevalence of the sickle cell trait, marriages occur between relatives as well as nonrelatives who are carriers hence consanguinity or nonconsanguinity may just have the same effect on occurrence [32].
The median age of diagnosis of sickle cell disease from our study was 7 days (IQR 7), this is at variance with an earlier report from Lagos, Nigeria that reported 27.33 (± 26.36) months [9]. The advantages newborn screening would provide for this community are; earlier age at diagnosis, early enrolment into comprehensive care that will result in a significant reduction in morbidity and mortality. A 15-fold reduction in deaths from sickle cell disease has been observed in countries that routinely screen newborns [26]. All babies with sickle cell anaemia in this study were enrolled in our hospital's haematology clinic for comprehensive care for sickle cell disease. Mothers who had babies with a sickle trait were also educated on its implications.
Universal newborn screening is required in Bida community, as more than a quarter of the newborn population have abnormal haemoglobin variants. The awareness of haemoglobin phenotype is low and the perception of haemoglobinopathies is widely wrong. Consanguinity though common in the population did not significantly affect the occurrence of sickle cell disease. A large-scale, multi-level health campaign would be needed to raise awareness that would stem the tide of haemoglobinopathies in the community.
It is recommended that the State Ministry of Health should implement newborn screening for haemoglobinopathy in this community so that comprehensive care can start early in affected neonates and public enlightenment campaigns about sickle cell disorders should be carried out through readily available media outlets. Education on sickle cell disorder can also be incorporated into the curriculum of primary and secondary schools to empower the younger generation with the knowledge required to make informed choices.
Abnormal haemoglobin variants were present in more than onequarter (26.4%) of the neonatal population studied in Bida. Most parents were not aware of their haemoglobin phenotype and had a wrong perception of sickle cell disease. Consanguinity though common in the population did not significantly affect the occurrence of an abnormal haemoglobin phenotype.
The extent to which the findings of this study can be generalized is limited by a relatively small sample size due to limited financial resources. If more funds were available it would have been desirable to collect data from more local government immunization centres across Niger State.
Considering the burden of sickle cell disorder and the carrier state in Bida, Niger State, it is apt that the State Ministry of health implements newborn screening for haemoglobinopathy in this community so that comprehensive care can start early in affected neonates.
Public enlightenment campaigns about sickle cell disorders should be done through the media, and at community meetings to ensure constant dissemination of information regarding the disorder, and to promote early testing.
Education on sickle cell disorder should be incorporated into the curriculum of primary/ secondary schools to enlighten adolescents and help them make informed choices about their life partners in future.
[Google Scholar] [PubMed]
[Google Scholar] [PubMed]
[Crossref]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
Citation: Folayan OS, Bello AO, Ernest SK (2023) Newborn Screening for Haemoglobinopathies in Bida, North-Central Nigeria. J Hematol Thrombo Dis. 11:537.
Received: 01-Mar-2023, Manuscript No. JHTD-23-21890; Editor assigned: 06-Mar-2023, Pre QC No. JHTD-23-21890 (PQ); Reviewed: 20-Mar-2023, QC No. JHTD-23-21890; Revised: 27-Mar-2023, Manuscript No. JHTD-23-21890 (R); Published: 03-Apr-2023 , DOI: 10.35248/2329-8790.23.11.537
Copyright: © 2023 Folayan OS, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.