Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2025)Volume 18, Issue 2
Background: As the malignant tumor with the highest incidence in the world, Breast Cancer (BC) is the number one killer of women's healthy life. Recently, tumor metabolic reprogramming has attracted extensive attention, and mitochondria play a vital role in this process as a metabolic hub. In this study, we developed a risk signature of Mitochondrial-Associated Genes (MRGs) and evaluated its ability to predict prognosis and risk stratification in BC patients.
Methods: The transcriptomic data and clinical features of BC samples were extracted from TCGA and METABRIC. We constructed an 8 MRG signature by LASSO combined with Cox regression and assessed the performance by ROC curve. Subsequently, we combined the risk scores based on the signature with clinical features to construct a nomogram model and evaluated its accuracy by clinical calibration curve and decision curve analysis. Functional enrichments and immune-related analyses were performed to compare the different status between high and low-risk groups. Finally, we compared the mutation landscape and drug sensitivity to explore the treatment response.
Results: A total of 8 MRGs (ACSL1, ALDH2, MTHFD2, MRPL13, TP53AIP1, SLC1A1, ME3 and BCL2A1) was constructed. This signature was an independent risk predictor for the survival of BC patients with a high hazard ratio (HR=3.028 95% CI 2.038-4.499). The low-risk group has a better prognosis, enhanced immune infiltration, significantly different mutation landscapes, and a more sensitive response to antitumor drugs, while the high-risk showed the opposite trend.
Conclusion: The MRG signature is a novel prognostic risk signature that can be used as a predictor for patient stratification in BC.
Breast cancer; Mitochondria; Risk signature; Prognosis
According to the latest statistics of GLOBOCAN 2020, 2.26 million new cases of female BC have been detected annually worldwide, which has overtaken lung cancer and ranks first among all malignant tumors regardless of sex [1]. At present, due to the development of imaging screening combined with surgical methods, advances in radiotherapy, and progress in therapeutic drugs, the prognosis of BC patients has been greatly improved [2-4]. Despite the systematic and abundant treatment options, it is necessary to adopt a tailored approach to elicit the best response due to the high heterogeneity of BC [5]. With the development of microarray and next-generation sequencing technology, polygenic testing can help clinicians in practice by providing auxiliary clinical features [6]. From PAM50 used for molecular typing of BC [7] to the 21-gene (Oncotype DX Breast Recurrence Score®) [8] and MammaPrint™ 70-gene signature [9] used to assess the clinical risk of BC patients, polygenic testing has gradually occupied an important position in guiding treatment.
As a central metabolic organelle, the mitochondrion performs critical functions for fatty acid, amino acid, and nucleotide metabolism in the development and progression of cancer [10]. At present, the fundamental effects of mitochondrial metabolism on steps of tumorigenesis, that is, malignant transformation, tumor progression, and therapeutic response, have finally been properly recognized [11,12]. Meanwhile, a variety of preparations specifically acting on mitochondria were developed according to the difference between cancer and normal cells [13,14]. In BC, targeting mitochondrial metabolism can provide a new vision for some subtypes with limited treatment options, such as Triple-Negative Breast Cancer (TNBC) [15,16]. Jiyoung Lee, et al., demonstrated that mitochondrial metabolism can be exploited to sensitize BC and potentially other tumor tissues to mitochondrial inhibitors [17]. In addition, Jui-Chih Chang, et al., revealed the antitumor potential of mitochondrial transplantation in BC cells via distinct regulation of mitochondrial function [18]. In other aspects, mitochondria are essential for the metabolism and activation of immune and cancer cells [19]. Furthermore, mitochondrial acquisition and increased oxidative phosphorylation have been proven to be involved in cancer progression and the development of chemotherapy resistance [20].
Given the existing findings, we constructed an 8 MRG signature by exploring BC samples in TCGA training cohort. ROC, Kaplan–Meier (K-M) survival analyses, nomogram model, Decision Curve Analysis (DCA) and calibration curve were used to assess the prediction ability and clinical application of the risk signature. The results indicate that our signature can effectively stratify risk populations and the 8 MRGs can serve as potential independent predictor in clinical application. Conclusively, our findings may provide new clues for the study of the mitochondrial metabolic targets of BC and lay a foundation for accurate immunotherapy targeting the mitochondrial metabolic pathway of BC.
Datasets and collection of mitochondria-related genes
The transcriptomic and corresponding survival data of TCGA samples were downloaded from the UCSC Xena database. The mutation and clinical features of BC were downloaded from TCGA by the “TCGAbiolinks” package. After removing the cases with missing data and male patients, we used TCGA sample barcodes to divide the cases into primary tumor (1036 cases) and normal tissues (99 cases). For verification, we also downloaded RNA-seq data from BC patients from the METABRIC database. After screening the cases, a total of 1903 patients were included in the analysis. By searching Molecular Signature Database v7.5 (MSigDB), we downloaded a MRG dataset (M9577) consisting of 450 genes. After integration with TCGA and METABRIC databases, a total of 418 MRGs were included in the subsequent analysis. Relevant grouping information and clinicopathological features were shown in Table 1.
| Characteristic | TCGA (n=1036) | METABRIC (n=1903) | |
| Age (mean (SD)) | 58.05 (12.93) | 61.09 (12.98) | |
| Status (%) | |||
| Alive | 888 (85.7) | 801 (42.1) | |
| Dead | 148 (14.3) | 1102 (57.9) | |
| PT (%) | Tumor size (%) | ||
| T1 | 269 (26.0) | ≤ 2 cm | 820 (43.5) |
| T2 | 600 (7.9) | >2 cm and ≤ 5cm | 968 (51.4) |
| T3 | 127 (12.3) | > 5 cm | 95 (5.0) |
| T4 | 37 (3.6) | ||
| Tx | 3 (0.3) | ||
| PN (%) | Positive nodes (%) | ||
| N0 | 485 (46.8) | 0 nodes | 992 (52.1) |
| N1 | 345 (33.3) | 1~3 nodes | 604 (31.7) |
| N2 | 117 (11.3) | 4~9 nodes | 204 (10.7) |
| N3 | 72 (6.9) | ≥ 10 nodes | 103 (5.4) |
| Nx | 17 (1.6) | ||
| PM (%) | |||
| M0 | 861 (83.1) | ||
| M1 | 27 (2.6) | ||
| Mx | 148 (14.3) | ||
| Stage (%) | Stage (%) | ||
| Stage I | 173 (16.8) | Stage I | 474 (33.8) |
| Stage II | 589 (57.1) | Stage II | 800 (57.1) |
| Stage III | 238 (23.1) | Stage III | 115 (8.2) |
| Stage IV | 19 (1.8) | Stage IV | 9 (0.6) |
| Stage X | 12 (1.2) | Stage 0 | 4 (0.3) |
| Subtype (%) | |||
| BRCA_LumA | 484 (46.7) | BRCA_LumA | 678 (35.6) |
| BRCA_LumB | 187 (18.1) | BRCA_LumB | 461 (24.2) |
| BRCA_Her2 | 72 (6.9) | BRCA_Her2 | 220 (11.6) |
| BRCA_Basal | 165 (15.9) | BRCA_Basal | 199 (10.5) |
| Unknown | 128 (12.4) | Unknown | 146 (7.7) |
| Claudin-low | 199 (10.5) | ||
|
Note: The TCGA cohort was used as training cohort and MERABRIC cohort as the validation cohort. |
|||
Table 1: Clinicopathological characteristics of the BC cases in TCGA and MERABRIC datasets.
Construction and clinical application of the risk signature
After differential gene analysis between tumor tissue and normal tissue with the “DESeq2” package (log2Fold Change >1, adjusted P value<0.05), a total of 64 Differentially Expressed Genes (DEGs) were screened out (39 upregulated and 25 downregulated). By performing univariate Cox regression After LASSO variable screening, an 8-MRG signature was established. The risk formula was as follows:
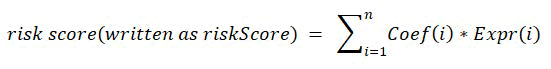
All patients were divided into high and low-risk groups based on the median riskScore. K-M plots was used for survival analysis. The predictive power of the riskScore was evaluated by ROC, time-dependent ROC, and Area Under the Curve (AUC).
In clinical application, the correlation between risk groups and clinical features was demonstrated by the “ggpubr” package. A nomogram model was constructed according to the Cox regression analyses. To assess the model’s predictive discriminatory power, we calculated the Concordance index (Cindex) and plotted DCA and calibration curves.
Differential gene and functional enrichment analyses between risk groups
To explore the causes underlying different prognosis, we performed differential gene and functional enrichment analyses between risk groups. Gene Ontology (GO) analysis was conducted to identify GO categories by their Biological Process (BP), Molecular Functions (MF), and Cellular Components (CC). Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was used to explore the influenced pathways among the risk groups. Gene Set Enrichment Analysis (GSEA) was performed to discover enriched gene sets between risk groups.
Immune related analyses
The estimation of stromal and immune cells in malignant tumor tissues using expression (ESTIMATE) data algorithm can infer the content of tumor cells as well as differentially infiltrating normal cells based on the unique nature of the transcriptional profile of cancer samples. By calculating the expression levels of the stromal signature and immune signature in BC patients, the related score of corresponding patients can be obtained. CIBERSORT was used to predict the proportion of 22 types of Tumor-Infiltrating Immune Cells (TIICs) in each tissue. The cells correlated with riskScore were presented as scatter plots with a significance threshold for P<0.05. Vesteinn Thorsson, et al., classified TCGA BC samples into different subtypes based on immune-related gene expression. The composition of immune subtypes between risk groups was presented by column graphs, and their differences were detected by the chi-square test processed by the “ImmuneSubtypeClassifier” package. In addition, single-sample GSEA (ssGSEA) allowed us to define enrichment scores that predict the abundance of 28 Tumor- Infiltrating Immune Cells (TIICs) in each sample based on a gene set of 782 genes.
Mutation landscape and Weighted Correlation Network Analysis (WGCNA)
After matching with TCGA barcodes, the top 15 most frequently mutated genes in different risk groups were displayed by oncoplots. We further examined somatic interactions, performed survival analysis and calculated Tumor Mutational Burden (TMB) according to risk groups. The above analysis and visualization were processed by the “maftools” package. Furthermore, we screened out module genes that were most strongly correlated with immune infiltration and risk level by the “WGCNA” package.
Treatment response and drug sensitivity
The effect of treatment on prognosis between different risk groups was demonstrated by K-M plots. Drug sensitivity training set data were downloaded from Genomics of Drug Sensitivity in Cancer (GDSC). Combined with TCGA data, we screened out commonly used antitumor drugs for analysis and presented the results through radar plots by the “oncoPredict” and “ggpubr” packages.
Statistical analysis
All statistical analyses and visualizations were conducted with R software (version 4.2.0), and the image editing and combination were completed by Adobe Illustrator. Student’s t test was used for statistical comparisons. Differences in gene expression and immune infiltration scores were detected by the Wilcoxon test, and discrepancies in riskScore between various clinical features were tested by the Kruskal Wallis test. K-M curves involved in the survival analysis were tested by log-rank test. The correlation plots were displayed by Spearman’s correlation test. Statistical significance was determined as a P value<0.05.
Preliminary screening of prognosis-related mitochondrial genes
The overall analysis process of this study is shown in Figure 1. A total of 418 MRGs were included for differential expression analysis. DEGs were defined as |log2FC|>1 with P value<0.05, and 64 Deferentially Expressed MRGs (DE-MRGs) were screened out. The distribution of all DE-MRGs is displayed in a heat map. After sorting by the P value of DE-MRGs, the top 5 genes in the up- and down regulated groups were selected to draw a volcano map. The 64 DE-MRGs were then subjected to univariate Cox regression in BC patients, and 9 significant OSrelated genes were detected (P<0.05) and presented in a forest plot (Figure 2).
Schematic flowchart of the workflow performed to build and validate the breast cancer prognostic risk signature. Some typical analysis results were shown in reduced size (normal-sized pictures are shown later).

Figure 1: The overall analysis workflow of this study.

Figure 2: Differential distribution of MRGs and profile plot of LASSO regression.
Note: (A) Differential distribution of MRGs between tumor and normal tissues in the TCGA cohort. (B) Volcano plot of the DEGs. (C) Univariate Cox regression identified MRGs significantly related to OS (P<0.05). (D) LASSO coefficient profile plots of the 8-MRG risk signature. (E) Penalty plot for the LASSO regression for the MRGs with error bars denoting the standard errors. MRG mitochondria-related genes, DEG differentially expressed gene, LASSO least absolute shrinkage and selection operator.
Construction of the risk gene signature by LASSO
9 OS-related DE-MRGs were included in the LASSO regression and finally 8 genes were constituted the MRG risk signature. The riskScore was calculated as follows:
riskScore=(0.153 × expr_ACSL1)+(0.022 × expr_ALDH2)+(0.029 × expr_MTHFD2)+(0.265 × expr_MRPL13)+(-0.417 × expr_TP53AIP1)+(-0.086 × expr_SLC1A1)+(-0.137 × expr_ME3)+(-0.302 × expr_BCL2A1).
The descriptions of the 8 MRGs are given in Table 2.
| Gene | Full name | Description |
| ACSL1 | Acyl-CoA Synthetase long-chain family member 1 | Exists at the outer mitochondrial membrane and plays an important role in ferroptosis in diverse cancer cell types. |
| ALDH2 | Aldehyde dehydrogenase 2 | Aldh2-deficient can activate a variety of carcinogenic pathways and promote the occurrence of hepatocellular carcinoma. |
| MTHFD2 | Methylenetetrahydrofolate dehydrogenase 2 | Regulates purine synthesis and signal transduction in activated T cells to promote proliferation and induces immune invasion of cancer cells by upregulating PD-L1. |
| MRPL13 | Mitochondrial ribosomal protein L13 | Exists in the mitochondria of eukaryotic cells, a poor prognostic factor for BC. |
| TP53AIP1 | Tumor protein P53 regulated apoptosis inducing protein 1 | TP53 target, plays a key role in inducing apoptosis in response to UV-induced DNA damage. |
| SLC1A1 | Solute carrier family 1 member 1 | An extracellular glutamine transporter, promotes tumor growth through reprogramming glutamine metabolism of natural killer T-cell lymphoma, while rendering tumor cells sensitive to asparaginase treatment. |
| ME3 | Malic enzyme 3 | Catalyzes oxidative decarboxylation of (S)-malate to pyruvate and reverse the decarboxylation reaction. Involved in the carcinogenesis of pancreatic cancer. |
| BCL2A1 | BCL2 related protein A1 | Bcl-2 family member, regulates endogenous apoptosis and target anti-apoptotic members. |
Table 2: Brief description of 8 MRGs.
Evaluation and expression pattern of the risk signature in the TCGA cohort
A heatmap was used to demonstrate the relationship between the 8 genes and clinical features after ranked the riskScore form low to high. The overall AUC was 0.647 (Figure 3B), while the predictive power of survival gradually increased over time, and the AUCs at 1 year, 3 years, 5 years, and 10 years were 0.58, 0.65, 0.67 and 0.76, respectively.
Subsequently, age, PT, PN, AJCC stage, and riskScore were included in the univariate Cox regression analysis. The Hazard Ratios (HRs) and P values are shown in a forest map. The results showed that all variables had a significant impact on prognosis and the risk score ranked the top: Age (HR=1.035, 95% CI: 1.021-1.049, P<0.001), PT (HR=1.559, 95% CI: 1.265-1.922, P<0.001), PN (HR=1.592, 95% CI: 1.338-1.894, P<0.001), stage (HR=2.192, 95% CI: 1.738-2.765, P<0.001) and risk score (HR=3.496, 95% CI: 2.364-5.171, P<0.001). Furthermore, the variables were incorporated into multivariate Cox regression. The results showed that age (HR=1.035, 95% CI: 1.021-1.049, P<0.001), stage (HR=1.863, 95% CI: 1.242-2.795, P=0.003), and risk score (HR=3.028, 95% CI: 2.038-4.499, P<0.001) were independent risk predictors for survival after adjusting for cofactors (Figure 3).
Among the 8 MRGs, 4 genes (ACSL1, ALDH2, TP53AIP1, and ME3) were downregulated in tumor tissues and upregulated in normal tissues, while another 4 genes (MTHFD2, MRPL13, SLC1A1, and BCL2A1) showed opposite trends (Figure 3). The boxplot was used to show the expression status of the MRGs between risk groups based on median risk score.

Figure 3: MRG risk signature and clinical application. (A) 8 MRGs and distribution heatmap of clinical features. (B-C) ROC and time-ROC analysis of the risk signature. (D-E) Univariate and multivariate Cox regression for the riskScore and corresponding clinical features. (F) 8 MRG expression levels between tumor and normal tissues. (The plot annotations were as follows: *if P<0.05, **if P<0.01, and ***if P<0.001 and ns if non-significant.)
Clinical translation of risk signature
The independent risk predictors (age, stage, risk score) were selected to construct a nomogram model. In terms of evaluation, the C-index was 0.763, and its standard error was 0.045. In addition, compared with single predictors, the DCA curve exhibited a higher net benefit based on the nomogram decision. In the calibration curve, the nomogram also showed an accurate ability to predict survival at 1 year, 3 years, and 5 years (Figure 4).

Figure 4: Construction and evaluation of the risk signature based nomogram model. (A) Age, stage, and riskScore (independent risk factors) were enrolled in the nomogram, and the total score could be used as a tool to predict the 1, 5, and 10-year prognosis of breast cancer patients. The orange dots and arrows in the plot represent the clinical features of a patient and the survival probability of the corresponding year. (B) DCA of the nomogram and single variable. (C) Calibration curve of the nomogram to predict survival at 1 year, 3 years, and 5 years. (D) KM plot for the prognosis analysis between high and low-risk patients. DCA decision curve analysis.
Validation and association of clinical features with risk score in TCGA and METABRIC
The prognosis between different risk groups was shown by K-M curve. The low-risk group showed a significantly better prognosis (P<0.001, Figure 4). The survival status of TCGA BC patients was displayed by a scatter plot based on risk score ranging from low to high. Correlations between risk score and PT, PN, AJCC stage, and PAM50 subtype are illustrated by boxplots. In the METABRIC validation cohort, survival analysis was also presented by K-M curves and scatter plots. Instead of PT and PN stage, the corresponding factors in METABRIC were displayed by tumor size and number of positive lymph nodes. Therefore, relevant variables were included to demonstrate in the boxplot. In addition, according to the median gene expression, the effect of these 8 MRGs on OS was shown by the K-M curve. The results showed that 3 gene reached statistical significance, in which high ME3 and TP53AIP1 expression was associated with a significantly better prognosis (ME3: P=0.013, TP53AIP1: P<0.0001), while high MRPL13 expression got a significantly worse prognosis (P=0.0017).
Functional enrichment analyses
To explore the underlying reasons for the prognosis between high and low-risk patients, we processed differentially expressed genes between risk groups and performed functional enrichment analyses among these DEGs. GO and KEGG pathway analyses showed that the risk DEGs were mainly associated with the regulation of T-cell activation, leukocytemediated immunity, and the NF-kappa B signaling pathway (Figure 5). Furthermore, GSEA table showed that the immune response and positive regulation of immune system processes were enriched in low-risk patients, while in high-risk patients, neuron differentiation and development were ranked the top.

Figure 5: Functional enrichment analyses of the different risk groups. (A-C) GO, KEGG, and GSEA analyses of the DEGs between the high and low-risk groups. GO Gene Ontology, KEGG Kyoto Encyclopedia of Genes and Genomes, GSEA Gene set enrichment analysis.
Assessment of the ability of the risk signature to distinguish different immune infiltrations
The ESTIMATE results showed that the low-risk patients exhibited significantly higher ESTIMATE, immune, and stromal scores and lower tumor purity (P<0.05, Figure 6). Subsequently, tumor-infiltrating immune cells were analyzed by CIBERSORT, and the cells significantly related to risk score were screened out from 22 types of immune cells. Moreover, the BC samples in the TCGA cohort were divided into 5 different immune subtypes: C1 (Wound Healing, 32%), C2 (IFN-gamma Dominant 51%), C3 (Inflammatory, 11%), C4 (Lymphocyte Depleted, 3%), and C6 (TGF-beta Dominant, 3%). The proportion of C1 was lower in the low-risk group, while the proportion of C3 and C6 was higher. In contrast, a higher proportion of C1 and a lower proportion of C3 and C6 were found in high-risk patients. In addition, the ssGSEA results showed that there was a significant difference in the enrichment of immune cells between the highrisk and low-risk groups, except for neutrophils (Figure 6).

Figure 6: Immune infiltration of the risk groups. (A) The ESTIMATE algorithm presented the immune infiltration scores across risk groups, including the ESTIMATE score, immune score, stromal score and tumor purity. (B) Immune subtype of TCGA breast cancer cases based on immune-related gene expression and the different subtype proportions in risk groups. (C) ssGSEA demonstrated the abundance of 28 tumorinfiltrating immune cells in individual tissue samples by a heatmap ranked from the lowest to the highest risk score. (The plot annotations were as follows: *if P<0.05, **if P<0.01, and ***if P <0.001 and ns if nonsignificant) ESTIMATE Estimation of Stromal and Immune cells in malignant tumor tissues using expression.
Risk signature and prognosis of different gene mutation landscapes
Patients in the risk groups were matched by sample barcodes, and the mutation landscape was displayed by oncoplots (Figure 7). In the combination of PIK3CA-associated genes affecting prognosis, CDH1 and KMT2C were the top 2 genes ranked by P value. In survival analysis, PIK3CA gene mutation (HR=2.03, P=0.0975) and co-mutation with CDH1 (HR=8.86, P<0.001) and KMT2C (HR=5.3, P=0.0138) in high-risk patients showed poor prognosis. However, the same genes did not reach significant difference in the low-risk group. Furthermore, the tumor mutational burden was calculated, and the TMB was 0.74/MB in high-risk patients and 0.58/MB in low-risk patients.

Figure 7: Mutation landscape analysis between risk signatures. (A-B) Oncoplots for the different mutation landscapes of risk groups. (C-E and G-I) K-M curve for the most frequently affected gene and combination of associated genes affecting prognosis in the high- and low-risk groups. (F, J) The calculated TMB in high and low-risk patients.
WGCNA of the risk DEGs
After WGCNA clustering, the overall situation between individual cases and ESTIMATE, riskscore, and risk group was shown by a sample dendrogram trait heatmap (Figure 8). Among the 3 modules, the turquoise module was positively correlated with the ESTIMATE, stromal, and immune scores but negatively correlated with tumor purity, higher risk score and high-risk group. The function of turquoise module was explored by GO analysis.

Figure 8: WGCNA, treatment responses and drug sensitivity. (A) Dendrogram trait heatmap for the overall situation between individual cases after WGCNA clustering and ESTIMATE, riskScore, and risk group. (B) Three color module genes clustered by WGCNA and corresponding statistical correlation. (C-E) KM plot for the prognostic analysis of patients with chemotherapy, radiotherapy and endocrinotherapy among different risk groups in the TCGA cohort. (F-G) Radar plots for drug sensitivity between different risk groups among IC50<1 and IC50>1.
Treatment response and drug sensitivity
After matching treatment information in TCGA and METABRIC, the results showed that the prognosis of highrisk patients in the three treatments was significantly worse (Figure 8). By screening common therapeutic drugs in clinical practice, the sensitivity of risk groups to drugs was analyzed. The results showed that the high risk was accompanied by a higher IC50, which means less sensitivity to drugs.
Reprogramming of metabolism has received strongly increasing attention within the last decade, and several studies have shown that altered mitochondrial metabolism is considered to be one of the major emerging mechanisms of therapeutic resistance. The construction of multi-gene prognostic risk prediction models by bioinformatics algorithms can help us to further discover the role of multi-genes in the diagnosis and treatment of BC. At present, Luo, et al., used the gene set of mitochondrial GOBP term combined with OS analysis to construct a gene prediction model, and preliminarily explored its correlation with immune infiltration. We selected 450 mitochondrial gene sets containing “mitochondr_HG-U133A_probes” with the systematic named M9577 by searching MSigDB. In terms of strategy, we first performed differential analysis between cancer and normal tissues, and then combined with LASSO-Cox algorithm, we constructed a risk signature consisting of 8 MRGs (ACSL1, ALDH2, MTHFD2, MRPL13, TP53AIP1, SLC1A1, ME3, and BCL2A1). Subsequently, we fitted a nomogram model based on this signature to test its clinical translation ability, and verified its risk stratification ability by immune infiltration analysis, risk group differential analysis, and drug sensitivity analysis.
In clinical translation, the total AUC was 0.647, and we calculated the AUC for 1, 3, 5, and 10 years, and its trend was gradually strengthened (from 0.58 to 0.76), which means that this signature also has a certain accuracy in the long-term prognosis of BC (Figure 3B-C). Cox regression analysis combined risk score and clinical features showed that risk score was also an independent risk factor affecting prognosis, and the HR ranked first among all factors (Figure 3D-E).
Given the excellent predictive power of the risk score, we constructed a nomogram combining the results of multivariate cox regression. DCA is a method for evaluating nomograms that can meet the practical needs of clinical decision-making, and a calibration curve is another method to evaluate the consistency between the predicted and true values. The above methods proved the effectiveness of the nomogram model based on the risk signature. In addition, the prognosis of high-risk patients was significantly worse, and a higher risk score was positively correlated with more aggressive clinical features. The corresponding results were also verified in the METABRIC validation cohort.
Among the 8 MRGs, three genes (ACSL1, MTHFD2 and MRPL13) were highly expressed in the high-risk group and tumor tissues. While TP53AIP1 and ME3 were highly expressed in the low-risk group and normal tissues. To be specific, the prognosis with high ME3 and TP53AIP1 was significantly better, while the prognosis with high MRPL13 was worse, it suggested that TP53AIP1 and ME3 may act as tumor suppressors and MRPL13 as an oncogene in BC. Similar to what we inferred, one study showed that TP53AIP1 has been proven to be a new tumor suppressor gene for BC and may become an effective target gene for therapy. Another study reported that high MRPL13 is a poor prognostic factor for BC, and it can be used as a molecular marker for prognosis judgment and as a potential therapeutic target. It is worth noting that MRGs are associated with the immune response and cell death, which also proves the reliability and potential to be used as biomarkers of our risk signature.
To explore the underlying reasons, we analyzed DEGs between risk groups and performed functional enrichment analyses. Most BP terms were related to the immune response, and KEGG analysis showed that Th17-cell differentiation, the NFkappa B signaling pathway and most terms were closely related to immune response and oncogenesis. Meanwhile, GSEA results also showed the up-regulation of several immune-related items in the low-risk group. These findings indicate that a low risk score may be accompanied by an abundant immune response. While in the high-risk group, the terms related to neuron differentiation ranked top, which may be related to the role of nerve growth factor in the proliferation, invasion and metastasis of BC cells, especially for TNBC.
The ESTIMATE algorithm can effectively evaluate the proportion of tumor, infiltrating immune, and stromal cells in the Tumor Microenvironment (TME). Similar to our findings, the low-risk patients exhibited higher scores of ESTIMATE, immune and stromal, while lower tumor purity, that is, presented a higher degree of immune infiltration and showed a “warmer” condition. Furthermore, CIBERSORT could evaluate the infiltration degree of 22 types of immune cells. Among them, the risk score was significantly negatively correlated with CD8 T cells, which are critical for tumor destruction, and the correlation coefficient was also the highest (R=-0.26, P<2.2e-10). Moreover, in M2 macrophages, which tend to promote angiogenesis and neovascularization, the risk score showed a significant positive correlation (R=0.19, P=2e-10). Notably, M2 macrophages can cause stromal activation and remodeling, which are endowed with a repertoire of tumor-promoting capabilities involving immunosuppression and result in poor prognosis of BC patients. Meanwhile, the risk score showed a negative correlation with M1 macrophages, which could enhance antitumor inflammatory reactions and act as major players in proinflammatory responses.
Meanwhile, based on multiple control modalities of intracellular and extracellular networks, TCGA cohort was divided into 5 subtypes (Figure 6B), in which C1 was higher in high-risk (38% vs. 26%), and C3 was higher in low-risk (16% vs. 7%). Among these subtypes, C1 (Wound Healing immune) has high expression of angiogenic genes and Th2 cells, which have high tumor cell proliferation and high intratumoral heterogeneity with a less favorable outcome. C3 (inflammatory), defined by elevated Th17 and Th1 genes, is associated with low to moderate tumor cell proliferation and minimal intratumoral heterogeneity, which has the best prognosis compared to other subtypes. Consistent with above, a low risk score was accompanied by increased immune cell infiltration, making the immune infiltration of low-risk patients look “warmer”. Therefore, it is reasonable to speculate that the risk signature can divide patients with different immune responses, and this heterogeneous status of tumor immunity may account for the difference in prognosis.
Moreover, we explored the mutational landscape between the risk groups. Among them, TP53 was the major mutation in high-risk patients (37%), and the incidence of PIK3CA mutation was dominant in low-risk patients (42%). TP53 mutation was associated with frequencies of mutations in TNBC and HER2- positive subtypes of 80% and 70%, respectively, and in luminal A and B types of 10% and 30%, respectively. PIK3CA mutation has been widely recognized as a genomic marker of BC, and PIK3CA mutation rates were lower in TNBC (16%) than in HR +/HER2-(42%) and HER2+(31%) BC. These findings partly explained the variation in riskScore among molecular subtypes. In addition, the same gene mutation status had a significantly worse prognosis in high-risk patients, which may be related to the overall worse prognosis of these patients. However, the higher TMB in high-risk patients may be due to the more TP53 mutations and the higher proportion of TNBC.
After WGCNA, it was found that the turquoise module gene trend was the most consistent with our speculation. GO analysis of the turquoise module genes also revealed that immune regulation may account for the differences. Moreover, the highrisk cohort had a worse prognosis in three treatments regardless of TCGA or METABRIC cohort, which also reflected the ability of our signature to screen for treatment response. The drug sensitivity analysis found that the low-risk group was more sensitive to drugs (such as paclitaxel, Docetaxel, Epirubicin and other commonly used chemotherapy regimens for BC), which may also account for the difference in the prognosis of chemotherapy patients.
In this work, we established an 8-MRG-based risk signature for BC prediction through joint analysis of gene expression datasets from TCGA and METABRIC, which also effectively stratified BC patients. The signature was constructed based on transcriptome data, and its feasible clinical translation ability was demonstrated by clinical validation methods. Moreover, this method might also be suitable to explore the prognostic effects of mitochondria-related genes in other malignant tumors. However, there are several limitations in our study. First, the risk signature was established based on prognosis, so its application is mainly reflected in the survival of BC patients and screening of high-risk populations but has little effect on early diagnosis and screening. Second, our analysis only used data from public databases, and more real-world data are needed to further confirm our findings. Third, although several basic experimental studies have validated the function of certain genes in our risk signature, the function of other genes and the underlying mechanism of these 8 MRGs need to be further investigated, and we will conduct corresponding experimental studies in the future.
In this study, we developed a novel 8-gene risk signature based on mitochondrial-related genes. Several clinical evaluation methods have confirmed the good prognostic accuracy of this signature. Importantly, our signature can effectively distinguish the risk populations among BC patients and has the potential to be used as a biomarker for clinical translation.
All data in our study are available upon reasonable request.
The authors declare that they have no competing interests.
This study was supported by Beijing Municipal Natural Science Foundation (No.7222145) and Shenzhen Key Medical Discipline Construction Fund (SZXK095).
We thank the investigators who participated and provided data unselfishly in the TCGA and METABRIC databases. It should be noted that an earlier version of this manuscript has been presented as Preprint.
The current study investigated publicly available data, and no ethical approval was needed. All methods were carried out in accordance with the Declaration of Helsinki.
All authors actively contributed to this manuscript, including the literature collection, and the design of the bioinformatics analysis. Yang Wang conceived the study design, performed the bioinformation analysis and wrote the manuscript. Ding-yuan Wang and Ke-na Bu conducted the literature collection and performed the statistical analyses. Yang Wang and Ding-yuan Wang collected the data and completed the figure illustration. Jidong Gao and Bai-lin Zhang reviewed the manuscript and made critical corrections. All authors commented on and revised previous versions of the manuscript. All authors read the final manuscript and approved the submission.
Consent for publication was obtained from all authors.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
Citation: Wang Y, Wang DY, Bu KN, Gao JD, Zhang BL (2025) Mitochondria-Related Gene Signature for Prognostic Prediction and Risk Stratification in Breast Cancer Patients. J Proteomics Bioinform. 18:690.
Received: 05-Oct-2023, Manuscript No. JPB-24-27329; Editor assigned: 09-Oct-2023, Pre QC No. JPB-24-27329 (PQ); Reviewed: 23-Oct-2023, QC No. JPB-24-27329; Revised: 03-Mar-2025, Manuscript No. JPB-24-27329 (R); Published: 10-Mar-2025 , DOI: 10.35248/0974-276X.25.18.690
Copyright: © 2025 Wang Y, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.