Journal of Physical Chemistry & Biophysics
Open Access
ISSN: 2161-0398
ISSN: 2161-0398
Review Article - (2014) Volume 4, Issue 2
Ion-radical mechanism of the phosphorylation in the three processes of paramount importance – enzymatic ATP synthesis, DNA replication, and enzymatic phosphorylation of proteins, which are three cornerstones of the life chemistry, was discovered by using pure isotope forms of metal (magnesium, zinc, calcium) ions catalyzing enzymatic phosphorylation. It is convincingly proved by magnetic isotope and magnetic field effects on these processes. Ionradical mechanism is inevitable because on the pathway of compression of reactants in any molecular machine (enzyme) electron transfer occurs which precedes generally accepted nucleophilic reaction. Ion-radical mechanism being controlled by magnetic interactions is switched on when at least two metal ions enter in catalytic site: the first one is tightly bound with phosphate group, the second one is ‘free’, and not bound with phosphate groups; it acts as an electron acceptor, it is a main actor of the ion-radical mechanism. This mechanism may be also switched off by presence of Fe ions. Ion-radical mechanism manifests itself in the ATP synthesis in isolated mitochondria and in the whole living organisms as well in the widely used polymerase chain reaction of the DNA replication. The mechanism can be used to stimulate ATP synthesis and eliminate ATP deficiency at cardiac diseases, to control cell proliferation, to kill cancer cells, and control trans-cranial magnetic stimulation against cognitive deceases.
<Chemistry is not the whole life but Life is totally chemistry. There are many factors controlling life-reproducing and life-supporting biochemical reactions, however the most intriguing is the magnetic field. The ability to respond to magnetic fields is ubiquitous and universal among the five kingdoms of organisms, [1] however a few areas of research are as controversial as biological and medical effects of magnetic fields.
An overwhelming majority of the biochemical reactions are those of phosphorylation, i.e. the transfer of phosphate group PO43-, individual or with attached molecular fragment (nucleotide, for instance). It is a key biochemical reaction; it plays a crucial role in the functioning of living organisms. Cell proliferation, gene expression, energy supply, the growth and reproduction of living organisms, the functioning of immune, mental and cell communication systems, physical mechanics and muscular contraction, metabolism − all these processes occur by phosphorylation of ADP, DNA and RNA chains, proteins and many other molecules in cell.
The heart of this paper is a new, recently discovered ionradical mechanism of the phosphorylation in the three processes of paramount importance – enzymatic ATP synthesis, DNA replication, and enzymatic phosphorylation of proteins – three cornerstones of the Life chemistry. The discovery of the ion-radical and, hence, magnetic field dependent mechanisms of phosphorylation forms a physically clear molecular concept as a key to understanding numerous enigmatic phenomena in magneto-biology and magneto-medicine.
Nucleophilic paradigm
In chemical biology it is established a solid viewpoint that enzymatic phosphorylation occurs as a nucleophilic reaction; it is generally accepted as a nucleophilic paradigm. It implies that the reaction proceeds as a unification of the two molecules, donor and acceptor of the phosphate group. Both donor and acceptor may be large, multiatomic molecules, however chemical transfer of the phosphate group itself involves very restricted numbers of atoms, not more than six. As a chemical event it may be presented as a consequence of the two elementary reactions. The first implies generation of the oxy-anion by deprotonating hydroxyl group: –OH → –O-+H+. Hydroxyl group may belong to inorganic phosphate (ATP synthesis by ATP synthase in mitochondria), to the phosphate group of donor (substrate ATP synthesis by phosphorylating kinases), to the ribose ring of the terminal group of the growing DNA chain (DNA synthesis and attachment of dNTP to the DNA), or to amino acid residue of peptides (phosphorylation of proteins).
The second step is accomplished as an attack of slightly positively charged phosphorus atom (its positive charge varies from +0.1 to +0.2 as follows from the calculations in terms of DFT theory) by oxy-anion:
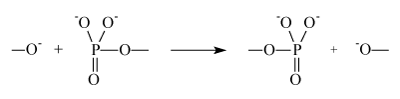
It is accompanied by transfer of phosphate group to oxy-anion synchronized with simultaneous release of the donor molecule residue. It is a key, universal step of any phosphorylation.
In order to accomplish nucleophilic reaction it is required to overcome repulsion between electron clouds of the reactants, i.e. repulsion induced by powerful exchange forces. It is so large that longdistance Coulomb interaction (attraction of reactants) in the transition state is not able to even partly compensate exchange repulsion. It means that the nucleophilic mechanism is energy strongly deficient. This undisputable physical argument has been proved by numerous calculations of the phosphorylation energy barriers on the basis of quite reliable methods of quantum chemistry including combined quantum chemistry/molecular dynamics techniques. Nucleophile transfer of phosphate group requires 42-46 kcal/mole; [2] nucleophile attack of hydroxyl to hydrolyze ATP needs to overcome an energy barrier 39 kcal/ mole [3]. A very high barrier for nucleophilic reaction is a reason why enzymatic phosphorylation does not occur in homogeneous solutions, it is accomplished only forcefully in enzymes, special molecular machines created by chemical Evolution. A source of energy needed to cover the energy deficit for energy-expensive phosphorylation is thought to be a compression of the catalytic site induced by protein mechanical energy; it compresses reactants overcoming exchange repulsion. (We will not discuss sources and molecular mechanics of the energy pumping; they are well known and beyond of this paper).
Enzymatic phosphorylation is known to be catalyzed by Zn2+, Ca2+ and Mg2+ ions; the latter dominate in vivo phosphorylation. The ions are traditionally considered to coordinate reactants in the catalytic site keeping them on the reaction trajectory to facilitate nucleophilic attack and probably slightly modify their reactivity via partial redistribution of charges in the reactants. There are two states of metal ion (suppose, Mg2+) inside of the catalytic site: one is tightly bound with phosphate group, the other is weakly bound, almost ‚free‘, coordinated mostly by water molecules. Namely this ion is supposed to be responsible for the catalysis of phosphorylation.
Electron transfer is inevitable step of the phosphorylation
The very important note: on the nucleophilic pathway of compression of reactants another reaction, electron transfer from the oxy-anion to the hydrated magnesium ion, occurs:
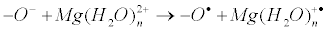
Being energy cheap (in contrast to energy expensive nucleophilic reaction) it is supposed to be switched on even at rather weak compression, i.e. electron transfer should inevitably precede nucleophilic reaction. The compression of catalytic site inevitably results in electron transfer which generates ion-radical pair and switches on a new, ionradical mechanism of enzymatic phosphorylation. Now we will discuss its functioning in the three cornerstone processes of phosphate group transfer; we will also discuss how ion-radical mechanism can be controlled by magnetic fields (both internal and external), how it may be switched on and off, and how it can be used in medicine.
Electron transfer in the ATP synthesis
ADP in the catalytic site is presented as a complex M (ADP)2+; we will specify M as Mg, Ca or Zn. Further the structure M (ADP)2+ is modeled by hydrated pyrophosphate complexes M (H2O)m2+(CH3P2O73-) with methyl group instead of an adenosine residue. Chart 1 illustrates their structure by examples of magnesium complexes in which Mg2+ ion is supposed to be coordinated to the oxygen atoms of the pyrophosphate anion and accepts two coordinating bonds. The other coordinating bonds may be used for addition of m water molecules, m being in the range from 0 to 4. The value m=4 corresponds to the fully completed six-coordinated shell of the tightly bound Mg2+ ion.
Chart 1: Magnesium complexes of the deprotonated (1) and protonated (2) pyrophosphates.
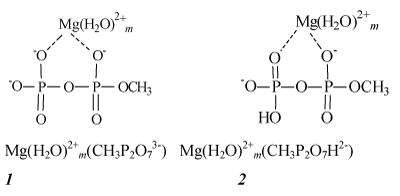
In complexes 1 pyrophosphate anions are deprotonated. However, in cells and mitochondria ADP is presented in both forms– deprotonated and partly protonated–almost equally, on a par, so that the structures of partly protonated complexes 2 should be also taken into account [4].
The energies of electron transfer from the complexes 1 and 2 (modeling ADP complexes) to the ‚free‘, hydrated magnesium ion, i.e. the energies of reactions
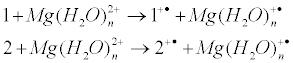
are shown in Figure 1 as a function of n, the number of water molecules in hydrated complex Mg(H2O)n2+. Note that the energy of electron transfer, being strongly dependent on n, is almost independent on m, the number of water molecules attached to the tightly bound metal ion.

Figure 1: Energies of electron transfer 1+Mg(H2O)n2+ → 1+• +Mg(H2O)n + (curve 1) and 2+Mg(H2O)n2+→ 2 +• +Mg(H2O)n +• (curve 2) as a function of the number of water molecules n in hydrated complex Mg(H2O)2+ n.
Figure 1 exhibits remarkable property. In water, i.e. at the conditions of the total hydration of magnesium ion (we will assume for this case n = ∞), electron transfer is endothermic, so that it is forbidden and ATP synthesis does not occur. When the value of n decreases and the ion becomes partly dehydrated, electron affinity of Mg(H2O)n2+ ion increases. For protonated complex 2 electron transfer becomes exothermic and energy allowed at n =3. For the protonated complex 1 electron transfer becomes exothermic even earlier, at n ≈ 12 (Figure 2).

Figure 2: Energies of electron transfer 1+Mg(H2O)n2+ →1+• +Mg(H2O)n +• as a function of n, the number of water molecules in the Mg(H2O)n 2+ ion.
The similar energy dependences of the electron transfer were detected for the calcium and zinc ions (for details see [5]).
The remarkable property of enzymes is that in the reactive state, when the enzyme domains are drawn together to unite substrate and ADP, they squeeze water molecules out of the catalytic site [6,7] and partly dehydrate M(H2O)n2+ ions. The removal of water molecules increases both positive charge q(M) on the metal ion and its electron affinity, i.e. electron transfer becomes exoergic and energy allowed, switching on a new, ion-radical mechanism of the ATP synthesis.
According to this mechanism, compression energy of enzymatic site is spent on the removal of water out of the ion hydrate shell which activates this ion as an electron acceptor. In order to make electron transfer allowed it is merely enough to remove weakly bound water out of the external hydrate shell with n ≥ 12 (for the deprotonated ADP) and n ≥ 4 (for the protonated ADP). In this process, a total energy deficit does not exceed 3–5 kcal/mol, i.e. it takes by order of magnitude less energy than nucleophilic ATP synthesis.
Electron transfer in the DNA synthesis
In this case electron transfer from the ribose oxy-anion to M(H2O)n2+ ion (M is Mg, Zn, Ca) switches on ion-radical mechanism of the DNA synthesis.
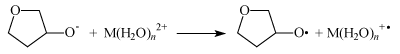
Like in the case of the ATP synthesis it is energy forbidden in water solutions when n = ∞ but it is seen in Figure 3 to be exoergic, energy allowed process even for ions with fully completed first hydrated sphere (n = 6).

Figure 3: Energy of the electron transfer as a function of n, the number of water molecules in M(H2O)n 2+ ions. M is Zn, Mg, and Ca. For n=1-6 the energies were computed, for n=∞ it is taken from reference 4.
Again, the removal of water molecules by compression of the catalytic site in DNA polymerases dehydrate ion and activates it as an electron acceptor switching on energy cheap ion-radical mechanism of the DNA synthesis.
Electron transfer in the protein phosphorylation
Electron transfer from the protein oxy-anion to the Mg(H2O)n2+ generates primary ion-radical pair [RO• Mg(H2O)n+•] composed of the oxy-radical RO• and hydrated radical-cation Mg(H2O)n+•:
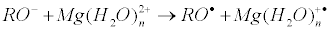
Here R is a protein residue; in particular, R is CH3 for threonine and Ph for tyrosine. The energies of these reactions were computed in terms of B3LYP theory. As shown in Figure 4 they are positive even for n < 12, i.e. these reactions are energy allowed.

Figure 4: Energy of the electron transfer for R=CH3 (1) and R=Ph (2) as a function of n, the number of water molecules in M(H2O)n 2+ ion.
Ion-radical mechanism of the ATP synthesis
An irrefutable reliability of the ion-radical mechanism is exhibited in magnetic isotope/magnetic field effects on the phosphorylation processes. We will start with the ATP synthesis. In experiments with creatine kinases (CK) which were loaded with isotope ions 24Mg2+, 25Mg2+, and 26Mg2+ it was shown that enzymatic activity of kinases with nonmagnetic isotope nuclei 24Mg and 26Mg were identical while kinase with magnetic nuclei 25Mg is almost twice more efficient (Figure 5). It is convincing evidence that magnetic isotope effect does occur in ATP synthesis while the classical, mass-dependent effect may be ignored [5].

Figure 5: The rate of ATP synthesis by CK as a function of magnesium isotopes. The rate A is given as radioactivity of 32P-ATP measured as a number of scintillations/min/mg of total amounts of protein (pure CK); concentration of MgCl2 is 15 mM.
Similar isotope effect is detected in CK loaded with isotope ions 40Ca2+ and 43Ca2+ (Figure 6); like in the case of magnesium ions enzymatic activity of CK with magnetic isotope nuclei 43Ca strongly exceeds that of CK with nonmagnetic nuclei 40Ca. At low concentration of CaCl2 there is no isotope effect; the largest effect is observed at [CaCl2] ≈ 120 mM [8] (As will be shown later in the concentration dependences of the ATP yield a very important information is hidden).

Figure 6: The rate of ATP synthesis by 40Ca-CK (1) and 43Ca-CK (2). A is the radioactivity of 32P-ATP (in scintillations/min/mg CK).
Mitochondrial CK with isotope zinc ions exhibits similar behavior [9] (Figure 7); many other examples of magnetic isotope effects on the ATP synthesis are given in [5].

Figure 7: The rates of ATP synthesis by CK in mitochondria as a function of zinc isotopes: 64ZnCl2 (1) and 67ZnCl2 (2). A is the radioactivity of ATP (in scintillations/min/mg of protein).
The ability of magnetic isotopes to stimulate ATP synthesis in the heart muscle of living rats was demonstrated by in vivo experiments. Injection of doxorubicin was to promote a significant damage to the local myocardial ATP synthesis suppressing it by about 70%. Then 24MgCl2 or 25MgCl2 was injected and after that a recovery of the ATP production up to the initial, pre-doxorubicin, level was observed. The extent of recovery as a function of magnesium concentration is shown in Figure 8. Evidently, there is a large isotope effect: delivery of 25Mg stimulates ATP synthesis by 2-3 times more efficiently than that of 24Mg. This is a first report on the isotope effect in a living organism manifesting its potential as an efficient remedy for the treatment of heart diseases [5].

Figure 8: The ATP production in living rats as a function of delivered amounts of Mg2+ ions.
Another irrefutable argument in favor of ion-radical mechanism is the effect of external magnetic field on the ATP yield [10]: in the Earth field, the rate of ATP synthesis by Mg-CK with 25Mg2+ ions is 2.5 times higher than that by enzymes with nonmagnetic nuclei 24Mg. The experimental results are in a perfect agreement with theoretical predictions [11,12].
Ion-radical mechanism formally imitates nucleophilic one and results to the same products; it includes a sequence of the following steps. The first one is an electron transfer from ADP to magnesium ion (AMP means adenosine monophosphate):
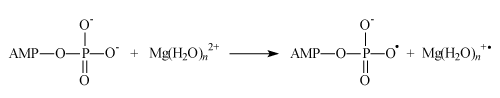
The second one is an attachment of the ADP radical to the inorganic phosphate (as it occurs in mitochondria):
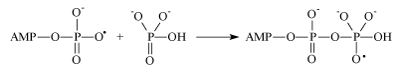
Generated in this reaction oxy-radical decomposes by well-known in chemistry β-scission reaction producing ATP and hydroxyl radical. Concluding reaction is a regeneration of Mg2+ ion by reverse electron transfer: Mg(H2O)n +• + •OH → Mg(H2O)n2+ + HO- nuclear spin dependence of ATP synthesis. As a first step the Scheme implies electron transfer from the terminal phosphate group of ADP to Mg2+ ion which generates primary ion-radical pair, composed of the radical-cation Mg+ and oxy-radical of ADP (reaction 1). Due to the total spin conservation this pair is in a singlet spin state. The next step is the phosphorylation itself in which the ADP oxy-radical attacks the P=O chemical bond of inorganic phosphate (reaction 2) generating new oxy-radical; the latter decomposes via β-scission of the P–O chemical bond (reaction 3) generating ATP and a final ion-radical pair (HO• Mg+•) which finally regenerates Mg2+ by back electron transfer. The rate of phosphorylation along the singlet channel (reactions 1-3 in the Scheme 1) is suppressed by spin allowed reverse electron transfer in the primary ion-radical pair which regenerates the starting reactants and decreases the ATP yield. However, in the presence of 25Mg2+, 43Ca2+ and 67Zn2+ ions, hyperfine coupling of the unpaired electron with the magnetic nuclei 25Mg, 43Ca and 67Zn in the Mg+, Ca+ and Zn+ ions stimulates singlet-triplet spin conversion of the primary ion-radical pair and transforms it into a triplet pair, in which back electron transfer is spin forbidden. This new triplet channel of phosphorylation (reactions 2’ and 3’) provides an additional yield of ATP which increases the total production of ATP by 2-3 times. The final ion-radical pair in the triplet channel undergoes fast triplet-singlet conversion due to electron spin relaxation and again regenerates Mg2+, Ca2+, and Zn2+ ions (for details see [5]).

Scheme 1: Generalizes this sequence introducing ion-radical pairs. It clearly illustrates
Ion-radical mechanism of the DNA synthesis
Nucleophilic mechanism of the nucleotide attachment to the growing DNA macromolecule implies direct attachment of 3´O− ion of the ribose ring to the Pα atom of the incoming nucleotide phosphate and simultaneous release of pyrophosphate anion [13,14]. Ion-radical mechanism suggests electron transfer from 3´O− ion to the Mg(H2O)n2+ ion as a key, primary reaction (Scheme 2); it generates primary ion-radical pair [3´O• Mg+•] composed of the oxy-radical 3´O• on the ribose ring and hydrated radical-cation Mg(H2O)n+•.

Scheme 2: Electron transfer generates ion-radical pair [3´O• Mg+•].
The second step is an addition of the ribose oxy-radical to the Pα−O double bond of the incoming nucleotide (Scheme 3).

Scheme 3: Attachment of the ribose oxy-radical to the nucleotide
It generates a new oxy-radical (further its simplified fragment will be denoted as OXY) which decomposes by β-scission mechanism along the three channels (Figure 9).

Figure 9: Schematic presentation of the three channels for the OXY ionradical decomposition.
Ion-radical mechanism imitates nucleophilic one [15,16]. The difference is that instead of one-step nucleophilic reaction (addition of 3´O− ion to the Pα atom of the incoming nucleotide, synchronized with the release of pyrophosphate anion) ion-radical mechanism implies three separate steps: electron transfer, addition of 3´O• radical to the incoming nucleotide phosphate generating new oxy-radical which mostly decomposes along the exoergic channels 2 and 3 without incorporation of nucleotide (channel 1 results in addition of nucleotide to DNA but it is endoergic and ignorable). For these reasons channels 2 and 3 dominate so that ion-radical mechanism appears to be strongly destructive and prevents DNA synthesis [17,18].
The source of the magnetic isotope/magnetic field effects is the primary ion-radical pair [3´O• Mg+]; namely this pair is responsible for the magnetic control of the DNA synthesis. As any pair generated from the diamagnetic molecules it is in a singlet spin state. In this pair spin allowed back electron transfer regenerates starting reactants; it makes ion-radical channel of the DNA synthesis inefficient. However, if magnesium ions with magnetic nuclei 25Mg are presented in the catalytic site, then singlet-triplet spin conversion, induced by hyperfine coupling in the ion-radicals 25Mg(H2O)n+•, successfully competes with back electron transfer and transforms short living singlet pair into the long living triplet pair, in which back electron transfer is spin forbidden. The increased lifetime of the triplet pair makes the addition of the ribose oxy-radical 3´O• to the Pα−O double bond more preferable. It generates secondary oxy-radical which mostly decomposes as described above. As a result, magnesium ions with magnetic nuclei (as well as Zn and Ca ions) stimulate singlet-triplet spin conversion of the primary ionradical pair and direct DNA synthesis along the ion-radical, destructive pathway suppressing ultimately DNA synthesis.
Figure 10 demonstrates convincing arguments in favor of this conclusion: both magnesium and zinc ions with magnetic nuclei strongly suppress DNA synthesis.

Figure 10: The rate of the DNA synthesis by polymerase β as a function of the magnesium and zinc ion concentration in pairs 24Mg2+/25Mg2+ (A), 26Mg2+/25Mg2+ (B), and 64Zn2+/67Zn2+ (C).
Polymerase chain reaction also exhibits magnetic isotope effect similar to that found for the pol β. Its magnitudes for magnesium and zinc ions are shown in Figure 11.

Figure 11: Isotope effects (w) on the PCR-induced DNA synthesis with magnetic (25Mg and 67Zn) and nonmagnetic (*Mg and *Zn) ions as a function of the ion concentration.
Both pol β and enzyme, producing PCR, demonstrate a common property: their activities are suppressed by 25Mg2+ and 67Zn ions in an approximately equal extent which depends on the concentration of ions. At low concentration isotope effect is negligible but it increases as the concentration increases. This phenomenon seems to be universal for the DNA synthesizing enzymes; at least, it is valid for the two different enzymes, pol and polymerase controlling PCR.
Figure 12 demonstrates magnetic field effect on the rate of DNA synthesis by pol β loaded with 24Mg2+ ions; the rate is slightly suppressed by magnetic field. On the contrary, magnetic field strongly, by 2-5 times, increases the rate of DNA synthesis by pol β loaded with 25Mg2+ ions. Both effects are physically clear substantiated [18].

Figure 12: Magnetic field effect on the rate of DNA synthesis by pol β loaded with 24Mg2+ (on the left) and 25Mg2+ (on the right) ions. Tritium radioactivity A is measured as the number of counts/min/mg DNA. Yellow (left) columns refer to the experiments in the Earth magnetic field; magenta (right) columns refer to the experiments carried out in magnetic field 160 mT.
Ion-radical mechanism of the protein phosphorylation
Phosphate group transfer from ATP to proteins induced by protein kinases should be also nuclear spin dependent, i.e. ion-radical mechanism of phosphorylation functions also in protein kinases. To confirm this statement we have studied phosphorylation of prothrombin catalyzed by prothrombin kinases with 24Mg2+ and 25Mg2+ ions. The yields of phosphorylated prothrombin as a function of MgCl2 concentration are plotted on the Figure 13. Like in the ATP synthesis by kinases, at the concentrations of MgCl2 less than 100 mM, there is no isotope dependence of the phosphorylation, however at higher concentration such a dependence clearly exhibits itself demonstrating higher efficiency of prothrombin kinase with 25Mg2+ ions in catalytic site in comparison with that of kinase with 24Mg2+ ions.

Figure 13: The yield A of phosphorylated prothrombin as a function of 25MgCl2 (1) and 24MgCl2 (2) concentration. A is measured as the radioactivity of 32P-prothrombin.
Nucleophilic mechanism of the protein phosphorylation implies direct attachment of the protein oxy-anion to the terminal phosphate group of ATP accompanied by release of ADP:
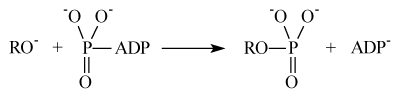
Ion-radical mechanism suggests electron transfer from the protein oxy-anion to the Mg(H2O)n2+ ion as a key, primary reaction:
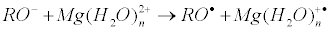
It generates primary ion-radical pair [–RO• Mg+•] composed of the oxy-radical RO• (remind that R is a protein residue) and hydrated radical-cation Mg(H2O)n+•. This reaction was shown before to be energy allowed even for n ≈ 12. Further oxy-radical RO• is attached to ATP generating unstable oxy-radical; its decomposition results in phosphorylated protein and ADP radical:
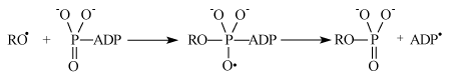
The latter generates ADP molecule and regenerates starting magnesium ion by electron transfer:
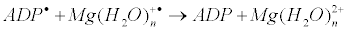
This mechanism is in accordance with isotope effect on the prothrombin phosphorylation (Figure 13).
How ion-radical mechanism is switched on?
First of all it is necessary to keep in mind that the concentration of ions indicated on the abscissa axes for the curves enzymatic activity versus ion concentration has nothing to do with real concentration of ions in the catalytic site; the latter may differ from average concentration in medium by many orders of magnitudes. Taking it into account one can easily understand a wide variety of relations between enzymatic activity and concentration of ions.
It is easy to see a general property of these dependences. At small concentrations of ions enzymatic activity increases as the ion concentration increases, no matter what ions are added, magnetic or nonmagnetic. At these concentrations all metal ions are tightly bound with phosphate groups; each site has at least one ion and the number of sites with a single ion increases as the concentration of ions increases. At these conditions only nucleophilic mechanism functions; the absence of magnetic isotope/magnetic field effects is a definite proof of this statement.
Further increasing of ion concentration results in appearance of the second metal ion in catalytic site; it is ‚free‘, not bound with phosphate groups. It is a main actor of the ion-radical mechanism, it acts as an electron acceptor (oxy-anions are donors, see above) and switches on ion-radical mechanism. Of course, both magnetic and nonmagnetic ‚free‘-metal ions function as an electron acceptor, both switch on ionradical mechanism; however the efficiency of magnetic ions strongly exceeds that of nonmagnetic ones. It is a reason of magnetic isotope effect; its appearance marks switching on ion-radical channel of enzymatic phosphorylation. It signifies a moment when at least two metal ions appear in the catalytic site of enzyme.
The boundary between two regimes of enzymatic phosphorylation, nucleophilic and ion-radical, depends on the enzyme and surrounding media. Thus, the boundary concentrations are <2, 2, and 80 mM for creatine kinase with Zn, Mg and Ca ions respectively (see Figures 5–7). For the protrombin kinase with Mg it lies about 100 mM (Figure 13); for the phosphoglycerate kinase with magnesium ion-radical mechanism is switched on at 10 mM. [19] Ion-radical mechanism of the DNA synthesis is switched on at ~ 0.5 mM for magnesium and at even less, <0.1 mM, for the zinc ions (Figure 10). These large differences characterize differences between concentrations of ions in enzymatic site and average ones in medium. They explain a great variety of concentration dependences exhibited by different enzymes.
It is worthy of noting that intra-cell concentration of ions in living organisms is supposed to be rather low and hardly overcome boundary when at least two ions happen to appear in enzymatic site. Perhaps it gives some grounds for thinking that in living organisms ion-radical mechanism is not dominating. However this argument is not universal; moreover, ion-radical mechanism may be artificially switched on for the medical purposes.
How ion-radical mechanism is switched off?
Recently ATP production by creatine kinase in the presence of magnesium isotopes was reexamined [20]. Neither isotope nor magnetic field effects were found in this reaction. However, the samples of MgCl2 used by the authors of 20 were strongly contaminated with Fe ions which were proved to destroy nuclear spin selectivity, delete magnetic isotope/magnetic field effects and suppress ATP synthesis. Figure 14 convincingly confirms this statement: the presence of FeCl2 even in concentration 10-2 mM is enough to eliminate nuclear spin/magnetic field effects. Moreover, it may be used to quantitatively separate contributions of nucleophilic and ion-radical mechanisms into the ATP production [21].

Figure 14: Isotope effect IE as a function of FeCl2 concentration (in mM; pay attention to the log scale for the latter).
This conclusion was directly confirmed by measuring ATP yield produced by two sorts of mitochondria loaded with 25MgCl2 and 24MgCl2 respectively [22]. Mitochondria were isolated from several rat tissues and the ATP yields were correlated with independently determined iron contents in these mitochondria. Isotope effect IE, that is the ratio of the ATP yields produced by mitochondria with 25MgCl2 and 24MgCl2 respectively, is different for different tissues (Figure 15). The mitochondria with high contents of Fe (spleen, liver) reveal no isotope effect (IE≈1.0), however mitochondria isolated from skeletal muscle, heart, kidneys and brain exhibit both low content of Fe and large isotope effect (IE≈1.8) in the ATP production. It immediately follows that the stimulation of the ATP synthesis by 25Mg2+ ions has a sense only in the tissues with low content of Fe in mitochondria. In particular, an outstanding promotion of the in vivo ATP synthesis in heart muscle (Figure 8) occurs due to the low iron content in the heart mitochondria.

Figure 15: Isotope effect IE in the ATP production by mitochondria from different tissues as a function of iron contents in these mitochondria. [Fe2+] is expressed in μg per g of mitochondria.
It is worthy to remind that on the reaction pathway the energy cheap ion-radical mechanism precedes energy expensive nucleophilic one. Being certainly unexpected, unpredictable, untraditional, and out-of-main-stream ion-radical paradigm meets a suspicious attitude. However, it should be taken into account that the ion-radical mechanism was discovered by using pure isotope forms of metal (magnesium, zinc, calcium) ions catalyzing ATP and DNA synthesis (the idea to use pure isotopes seemed to be mad and had occurred to nobody before to test it) and convincingly proved by magnetic field effects on these processes. Moreover, ion-radical mechanism manifests itself in the ATP synthesis in isolated mitochondria and in the whole living organisms as well in the widely used and popular polymerase chain reaction of the DNA replication. It is clear also how this mechanism can be used to stimulate ATP synthesis and eliminate ATP deficiency at cardiac diseases and how to use magnetic isotope ions as the medical agents against hypoxia and cardiac insufficiency, as a means for controlling cell proliferation and stimulating destruction and apoptosis of the cancer cells. Moreover, it is certainly known how ion-radical mechanism can be switched on or off by using magnetic isotope or iron ions respectively. It is also known in which organs of the living body this mechanism is the most efficient. No doubts ion-radical mechanism is expected should function in synthesis of GTP, CTP, and TTP as well as in synthesis of RNA. At last, it is unambiguously shown to have nothing to do with hazardous contaminations or impurities [5,18].
The molecular, ion-radical concept in magneto-biology seems to be most reasonable and significant for explaining biomedical effects of electromagnetic fields, for the progress of the new transcranial magnetic stimulation of cognitive activity, for the nuclear magnetic control of biochemical processes, and for the search of new magnetic effects in biology and medicine. The key structural element of the concept is an ion-radical pair as the receiver of magnetic fields and the source of magnetic effects. The existence of such pairs was convincingly proved in enzymatic ATP and DNA syntheses and in protein phosphorylation.
The discovery of the ion-radical and, hence, magnetic field dependent mechanisms of ATP synthesis, DNA replication and protein phosphorylation forms a physically clear molecular chemical concept in the magneto-biology as the base for its further development, as a means to control and predict new phenomena in magneto-biology. These three processes play a crucial role in the functioning of living organisms: in cell division, in the operation of the genetic apparatus, in gene expression and synthesis of the encoded enzymes, in control of biological clocks, in functioning of signaling system, etc. Ionradical mechanism is a key to understanding numerous phenomena of magneto-biology and medicine. Moreover, it prompts ideas for the new quests along the route of transformation of magneto-biology into a respected science. External magnetic field as a factor controlling reaction rates are important for trans-cranial magnetic stimulation, but internal (nuclear) magnetic fields are even more important: they produce large effects, easily accessible for practices by using stable magnetic isotope ions instead of those with natural isotope abundance [23].