Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Review Article - (2016) Volume 5, Issue 4
Ischemia-reperfusion injury is a common feature of ischemic stroke, which occurs when blood supply is restored after a period of ischemia. Reperfusion can be achieved either by thrombolysis using thrombolytic reagents such as tissue plasminogen activator (tPA), or through mechanical removal of thrombi. Spontaneous reperfusion also occurs after ischemic stroke. However, despite the beneficial effect of restored oxygen supply by reperfusion, it also causes deleterious effect compared with permanent ischemia. With the recent advancement in endovascular therapy including thrombectomy and thrombus disruption, reperfusion-injury has become an increasingly critical challenge in stroke treatment. It is therefore of extreme importance to understand the mechanisms of ischemia-reperfusion injury in the brain in order to develop effective therapeutics. Accumulating experimental evidence have suggested that the mechanisms of ischemia-reperfusion injury include oxidative stress, leukocyte infiltration, platelet adhesion and aggregation, complement activation, mitochondrial mediated mechanisms, and blood-brain-barrier (BBB) disruption, which altogether ultimately lead to edema or hemorrhagic transformation (HT) in the brain. Potential therapeutic strategies against ischemiareperfusion injury may be developed targeting these mechanisms. In this review, we briefly discuss the pathophysiology and cellular and molecular mechanisms of cerebral ischemia-reperfusion injury, and potential therapeutic strategies.
<Keywords: Stroke; Ischemia; Reperfusion; Oxidative stress; Leukocyte; Platelet aggregation; Complement; Mitochondria; Bloodbrain- barrier; Post-conditioning
Stroke, including ischemic stroke and hemorrhagic stroke, is a leading cause of death and disability world-wide. Cerebral ischemia is a major type of stroke, accounting for almost 80% of stroke cases [1]. Restoration of blood supply, referred to as “reperfusion”, is virtually a desired goal for acute stroke treatment. On the other hand, spontaneous reperfusion occurs commonly after stroke, in about 50-70% of ischemic stroke patients [2]. Reperfusion can also be achieved by thrombolytic therapy or endovascular therapy including thrombectomy using retrieval devices and thrombus disruption using stents. Intravenous administration of recombinant tissue plasminogen activator (r-tPA) is to date the only FDA-approved thrombolytic therapy for acute ischemic stroke treatment. tPA was originally demonstrated to be safe within 3 hrs after stroke onset, whereas recent studies have extended the time window of tPA thrombolysis treatment to 4.5 hrs after stroke onset [3]. In the past few years, a series of clinical trials for embolectomy surgery using stent retrievers have been completed in US and the overall outcomes are beneficial, therefore this treatment strategy is expected to be widely practiced in clinic in the near future [4]. However, despite the beneficial effect of oxygen restoration by reperfusion, rapid reperfusion causes deleterious effect for brain function, the so called “reperfusion injury”, which significantly worsens the functional outcomes of stroke. During the past two decades since recombinant tPA was approved for thrombolysis in stroke in North America in 1996 [5], the deleterious effect of reperfusion injury after ischemic stroke has been clearly demonstrated from both experimental studies and clinical evidence. For example, in a rat model of stroke using middle cerebral artery occlusion (MCAO), the infarct volume significantly increased at 6-24 hrs following the start of reperfusion compared to permanent occlusion, which is a solid confirmation of reperfusion injury following ischemia [6]. In addition, some MRI studies using fluid-attenuated inversion recovery and perfusion-weighted images for human stroke have shown that reperfusion could be linked to an early opening of the blood-brain barrier and consequently to secondary reperfusion injury and poor outcome [7]. Based on these, an ideal therapeutic strategy would be to restore oxygen supply to affected tissues, and in the meantime to minimize the reperfusion injury. So far there have been no therapies targeting reperfusion injury, it is therefore of extreme importance to understand the mechanisms of ischemia-reperfusion injury in the brain in order to develop effective therapeutics. Here in this review, we briefly discuss the pathophysiological mechanisms of reperfusion injury and potential therapeutic strategies.
Since the approval of intravenous administration of r-tPA for acute stroke treatment, the deleterious effect of rapid reperfusion on brain function has been widely recognized, and the underlying mechanisms of reperfusion injury have been revealed by accumulating clinical and experimental studies, part of which are learnt from ischemiareperfusion injuries in other organs such as heart and liver. The main mechanisms of reperfusion injury include oxidative stress, leukocyte infiltration, mitochondrial mechanisms, platelet activation and aggregation, complement activation, and blood-brain-barrier (BBB) disruption, which ultimately lead to brain edema or hemorrhagic transformation and eventually causing significant neuron death and neurological dysfunctions [8].
Oxidative stress in ischemia-reperfusion injury
Oxidative stress occurs when the manifestation of reactive oxygen species (ROS), mainly peroxides and free radicals such as superoxide anion, overwhelms antioxidant capacity. Oxidative stress is an important mechanism implicated in the pathogenesis and disease progression of many diseases including cerebral ischemia-reperfusion injury. Overproduced ROS cause direct damages to all cellular components including proteins, DNA, RNA and lipids. Increased superoxide generation has been detected in vivo in a cerebral ischemia-reperfusion model using cytochrome c-coated platinized carbon electrode, where the superoxide reduces immobilized ferric cytochromes C, then the latter is immediately re-oxidized by the electrode [9]. In another study, enhanced ROS production was detected using electron spin resonance (ESR) spectroscopy in the rat brain after cerebral ischemia-reperfusion [10]. Furthermore, oxidative stress-mediated BBB dysfunction has been evidenced in a cerebral ischemia-reperfusion model using superoxide dismutase (SOD)-deficient mice [11].
One major source of ROS production after ischemia-reperfusion is mitochondria. It has been established that ischemia-reperfusion induces post-translational modification of oxidative phosphorylation proteins (OxPhos), which increases mitochondria membrane potential (MMP), a condition that leads to excessive generation of ROS [12]. Another important source of ROS production is NADPH oxidase (NOX), a key component of electron transport chain in the plasma membrane, which generates free radicals by transferring one electron to molecular oxygen [13]. Experimental studies demonstrated that NOX2 gene deletion reduces BBB dysfunction and infarct size after filament MCAO for 2 h followed by reperfusion in mice [14], indicating the contribution of NOX in the pathogenesis of ischemia-reperfusion injury, and the potential of targeting NOX as a therapeutic strategy [15].
Although a large array of animal studies have established that oxidative stress is an important mechanism of reperfusion-injury, the direct evidence in clinical stroke patients is scant. For example, in a clinical study for stroke patients who had received tPA recanalization treatment, Domínguez et al. [16] found the oxidative stress markers, including malondialdehyde and myeloperoxidase, were already increased at baseline of stroke, whereas no further increases of these markers were found after tPA recanalization. This data raises concern arguing against the relationship between oxidative stress and reperfusion injury. However, this discrepancy could be due to the limitations of clinical investigation, for example, the peak of oxidative stress might have been missed. More carefully designed clinical investigation is warranted to further elucidate the roles of oxidative stress in ischemia-reperfusion injury.
Mitochondrial mechanisms in ischemia-reperfusion injury
Mitochondria are multi-functional organelles playing essential roles in physiological and pathophysiological conditions. In the context of ischemia-reperfusion injury, mitochondria are not only ROS generators, but also mediate other pathological processes such as apoptosis and necrosis [17,18]. It has been demonstrated that reperfusion after a long period of ischemia can result in the opening of mitochondrial permeability transition pore (mPTP), a non-specific pore in the inner mitochondrial membrane, which leads to Ca2+ flux into mitochondria, and subsequent mitochondria swelling and uncoupling and eventually ATP deprivation and cell necrosis [18]. In support of this mitochondria-mediated mechanism in ischemiareperfusion injury, a series of studies have shown that mPTP inhibitors such as CsA (cyclosporin A) or SfA (sanglifehrin A) confer protection for the brain against reperfusion-injury [18,19], which could be a potential therapeutic strategy.
Another important biological feature of mitochondria is their dynamics, mainly fission (division) and fusion, which is reflected by the dynamic combination of fragmented and tubular morphology of mitochondria [20-22]. There are many molecular players regulating mitochondria fission and fusion. Briefly, fission is mainly regulated by a member of dynamin family of guanosine triphosphatases (GTPases), which is Dynamin-like protein (Dnm1) in yeast and Dynamin-related protein 1 (Drp1) in mammals [23]. Mitochondria fusion begins with the contact and merge of mitochondrial outer membranes, which is mediated by dynamin family members mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) in mammals, whereas fusion between the inner membranes is mediated by another dynamin family member Opa1 in mammals [24]. A large volume of studies have suggested that fission and fusion are critical regulators for mitochondrial homeostasis (including turnover, maintenance and biogenesis), and protection and exchange of mitochondrial materials [25,26]. More importantly, emerging experimental evidence suggest that these dynamic morphological changes of mitochondria are implicated in cell survival and apoptosis, although the precise relationship remain not fully resolved. For example, it has been reported that Drp1 translocation is associated with mitochondrial fragmentation during apoptosis [27,28]. Moreover, Drp1 and Mfn2 are proposed to co-localize with Bax, a pro-apototic Bcl-2 family member, to form a fission complex [29,30]. These findings strongly support the involvement of mitochondrial fission and fusion in ischemia-reperfusion-induced cell injury and apoptosis. Indeed, some studies have reported that ischemia-reperfusion is accompanied by mitochondrial fragmentation [31,32]. These evidence also imply a potential strategy targeting mitochondria fission and fusion for ischemia-reperfusion treatment, which will be further discussed in section 3 of this review.
Leucocyte infiltration in ischemia-reperfusion injury
Inflammation plays a central role in the pathogenesis of cerebral ischemia-reperfusion injury, wherein leucocytes infiltration is a key initiating process. During reperfusion, activated leucocytes attach to endothelial cells through chemotactic signals, followed by the production of matrix metalloproteinase and neutrophil-derived oxidant, causing the disruption of blood-brain barrier. The leukocytes in turn extravasate from capillaries and infiltrate into brain tissue, releasing cytokines that mediate inflammation, which eventually result in deterioration of penumbra [33,34].
A series of animal studies have demonstrated the deleterious effect caused by leucocytes infiltration in ischemia-reperfusion injury. For example, in a rat stroke model of MCAO, Zhang et al. [6] found that neutrophil accumulation at the site of neuronal injury occurred earlier and to a greater extent in the reperfused tissue than in tissue with permanent occlusion. Furthermore, the contribution of leukocytes infiltration in reperfusion injury is also supported by the beneficial effects of neutrophil depletion, in which the animals after transient ischemia showed smaller infarct size when administered with antineutrophil antiserum or monoclonal antibodies [35,36]. Reperfusion injury can eventually cause BBB disruption and hemorrhagic transformation (HT). Some direct evidence indicates the involvement of leucocytes infiltration in HT, further supporting the roles of leucocytes infiltration in reperfusion-injury. For example, it has been reported that white blood cell count was significantly higher in stroke patients after receiving thrombolysis therapy with HT than those without [37]. The subsequent enhanced leukocyte infiltration may further enhance the damage of microvascular endothelial cells, and exacerbate BBB dysfunction.
Leucocyte infiltration is a cascade of processes including leukocytes migration directed by chemotactic signals, leukocytes “rolling” on the endothelium, leucocyte adhesion to the microvascular endothelial surface through receptor/ligands interaction, matrix metalloproteinase production for breakdown of BBB, leukocyte extravasation into brain tissue and finally the release of cytokines to brain tissue triggering inflammation response [38]. The molecular regulation mechanisms of leukocytes infiltration have been elucidated by enormous experimental studies. One important regulator of leukocytes rolling to endothelium is endothelial P-selectin, which is up-regulated by superoxide free radicals produced during ischemia and reperfusion. P-selectin interacts with its receptor on leukocyte, P-selectin glycoprotein Ligand- 1(PSGL-1), which facilitates the low affinity “rolling” of leukocytes on the endothelium [39]. Firm adherence of leucocytes to endothelium is mediated by the interaction of leukocyte β2 integrins CD11a/CD18 and CD11b/CD18 with endothelial intercellular adhesion molecule 1 (ICAM-1) [40]. The subsequent transmigration of leukocytes is regulated by platelet-endothelial cell adhesion molecule-1 (PECAM-1) along the endothelial cell junction [41]. Moreover, alpha-catenin and VE-cadherin, which form a complex, are also required for efficient transendothelial migration of leukocytes [42]. These important regulators for leukocyte infiltration may serve as potential targets for developing therapeutic strategies for ischemia-reperfusion injury treatment.
Experimental evidence have implied an important role of platelets in the pathogenesis of ischemia-reperfusion injury. It has been observed that platelets are activated by ischemia-reperfusion and accumulate in vascular beds at early time points after reperfusion [43]. Upon activation, platelets generate oxygen radicals and release proinflammatory factors such as platelet-derived growth factor (PDGF), arachidonic acid metabolites, thromboxane A2, serotonine, and platelet factor 4 (PF4). Mechanistically, a lot of studies have shown that the accumulation of activated platelets on endothelial cells and their interaction is mediated by P-selectin [44]. Another important player in platelet activation after reperfusion is platelet-activating factor (PAF), which has been suggested to play deleterious roles in various models of brain ischemia, evidenced by the beneficial effect of PAF antagonists in brain function [45]. PAF production in the brain was decreased after prolonged ischemia, but upregulated after reperfusion, suggesting its distinct role in reperfusion injury [46]. Moreover, a very recent study shows that platelet-activating factor receptor (PAFR) also plays crucial roles in global ischemia-reperfusion injury, as PAFR deficient mice exhibited significantly decreased brain infarct and improved neurological outcomes after ischemia-reperfusion [47].
Interestingly, platelet activation is also involved in leukocyte infiltration, as activated platelets adhere to microvascular endothelial cells, causing the latter to release mediators that results in chemotaxis and migration of leukocytes, thus exacerbating the inflammatory cascade [48]. The function of platelets in modulating leukocyte functional responses is potentially through the interaction between plateletsreleased fibrinogen and CD11C/CD18 [49]. Conversely, activated leukocytes can alter platelet function [50]. Indeed, emerging evidence have supported that platelets and neutrophils act synergistically in the pathogenesis of reperfusion injury, which is largely due to cell-to-cell interactions mediated by P-selectin [51].
Complement-mediated ischemia-reperfusion injury
As part of the innate immune system, the complement system consists of multiple cascades that all together play integrated roles in the initiation and regulation of inflammatory responses induced by pathogens or other stimuli. There are compelling evidence implying that the activation of complement system is one of the important mechanisms of cerebral ischemia-reperfusion injury [52]. Experimental studies suggest that complement system plays a direct role in neuronal cell death in the setting of CNS inflammation [53,54]. It has been demonstrated in a transient stroke model, 45 min of MCAO followed by 23 hr of reperfusion in mice, C1q gene expression is significantly up-regulated in neurons in the ipsilateral cerebral cortex, but barely detectable on contralateral side or nonischemic control brain [55]. This study provides evidence for the upregulation of early complement components in the setting of cerebral ischemia-reperfusion injury. The pathophysiological effects of C1q on ischemic neurons was further validated by administration of soluble complement receptor-1 (sCR1), a potent inhibitor of early complement activation, which inhibited neutrophil and platelet accumulation and reduced brain infarct size in a transient stroke model with 45 min of MCAO followed by 23 hours of reperfusion [56]. C1q therefore could be a potential therapeutic target for treatment of cerebral ischemiareperfusion injury. In another rat model of transient global ischemia, C1q expression was demonstrated to be up-regulated in microglial cells after 72 hours of ischemia at both mRNA and protein levels, using in situ hybridization and immuno-staining, respectively. Intriguingly, immuno-staining data in another study showed that C1q protein was exclusively expressed on microglial cells throughout all brain areas and at all-time points following ischemia-reperfusion injury [56]. Although the pathophysiological significance of C1q up-regulation in microglia after ischemia-reperfusion remains not resolved, the authors propose that C1q up-regulation may promote CNS inflammation through local complement activation, or C1q receptor binding and activation of inflammatory cells.
During reperfusion the complement system can be activated through different pathways, including the antibody-dependent classical pathway, the alternative pathway, or the letin pathway involving MBL/MASP (mannan-binding lectin/mannan-binding lectin-associated serine proteases) [57,58]. The classical, alternative and lectin pathways are initiated by C1q, MBL/ficolins/Collectin-11 and C3b, respectively, but all leading to the activation and cleavage of C3 into C3a and C3b. As a result, multiple inflammation mediators will be induced, including anaphylatoxins C5a, and the distal complement component C5b-9, which forms a complex called membrane attack complex (MAC) with another four complex proteins, C6, C7, C8, C9 [57]. C5a can stimulate leucocytes infiltration into damaged tissue, and may also induce the release of pro-inflammatory factor such as IL-1, IL-6 and TNFα. In addition, MAC forms transmembrane channels, which increase cell membrane permeability and eventually disrupt the phospholipid bilayers of cellular membrane, leading to cell lysis and death. Furthermore, MAC plays an essential role in mediating the recruitment of leukocytes to the reperfused tissue, potentially via local induction of IL-8 [59,60]. MAC also induces endothelial expression of monocyte chemo-attractant protein-1 (MCP-1), also knows chemokine (C-C motif) ligand 2 (CCL2), which is a critical player in inflammation responses in central nervous system disorders.
BBB dysfunction in ischemia-reperfusion injury
BBB is a selective permeability barrier that separates blood circulation from the brain tissue. It is mainly composed of endothelial cells, pericytes and astrocytes. BBB disruption is a common pathophysiological feature in cerebral ischemia-reperfusion injury, and to some extent can be considered an outcome of all the aforementioned ischemia-reperfusion injury mechanisms including oxidative stress, leucocytes infiltration, platelets activation and complement activation. It has been proposed that there are 3 stages in BBB permeability changes after ischemia-reperfusion. Stage 1 is reactive hyperemia, which is characterized by the loss of cerebral autoregulation and increased BBB permeability. Stage 2 is hypoperfusion caused by microvascular obstruction via swelling of endothelial cells and astrocytic end-feet, and formation of endothelial microvilli. This leads to nutritional deficiency in the brain tissue and enhances neutrophil infiltration and subsequent inflammation response. Stage 3 is the increase of paracellular permeability, which occurs as a biphasic response. The first phase occurs 3-8 hours post-reperfusion caused by enhanced inflammation and oxidative stress on the endothelial cells, accompanied by ECM degradation. The second phase occurs 18-96 hours post-reperfusion and coincides with the increased vasogenic edema and angiogenesis [61]. Stage 1, hyperemia, is associated with cytotoxic edema, while the biphasic stage 3 is characterized by vasogenic edema and altered BBB tight junctions, which ultimately lead to increased permeability for macromolecules across BBB. Figure 1 shows the schematic description of BBB changes after ischemia-reperfusion [61].
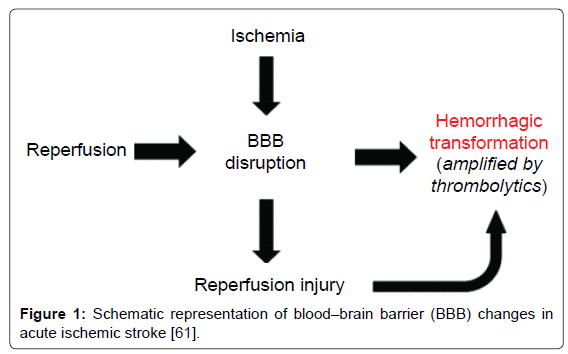
Figure 1: Schematic representation of blood–brain barrier (BBB) changes in acute ischemic stroke [61].
Both animal studies and clinical investigation have provided compelling evidence that BBB disruption occurs during cerebral reperfusion and can lead to vasogenic edema and hemorrhagic transformation. For example, Yang et al. [62] reported a BBB disruption after 3 hours of temporary occlusion followed by 3 hours of reperfusion, whereas the BBB remained intact after 6 hr-occlusion in a permanent occlusion group. In a clinical study for 144 acute focal stroke patients, Latour et al. [63] investigated the association between reperfusion, HT and clinical outcome. They found that BBB disruption was more common in patients with reperfusion (45%) than those without reperfusion (18%). Additionally, in the reperfused group, patients with BBB disruption were more likely to have a poor neurological behavior outcome (63%) than those without BBB disruption (25%). This is a critical study that established the association of early BBB disruption with reperfusion, and with HT and poor clinical outcomes in humans.
Potential Therapeutic Strategies Targeting Ischemiareperfusion Injury
It remains a big challenge for development of effective therapeutics against ischemia-reperfusion injury in the brain. With the underlying mechanisms of ischemia-reperfusion injury being revealed in the past few decades, an increasing number of potential strategies are being proposed and developed to limit or rescue ischemic-reperfusioninduced brain injury, targeting these different mechanisms.
Antioxidant treatment
Numerous experimental studies have shown that therapeutics using antioxidants such as iron-chelating compounds, catalase, superoxide dismutase, and vitamin E were able to prevent or attenuate ischemiareperfusion injury. For example, treatment with the iron-chelating compound deferoxamine was shown to be protective against ischemiareperfusion in newborn piglets [64]. Moreover, in a study using temporary MCAO model in rats, post-injury treatment with either recombinant Cu/Zn superoxide dismutase (SOD-1) or SOD-1 hybrid protein, SOD:Tet451, that is linked to a neuronal binding fragment, has significantly reduced brain infarct volume, and the neuronal targeting of SOD:Tet451 exerts more enhanced neuroprotective effect compared to SOD [65]. This SOD-based strategy was further improved by nanoparticle (NP)-mediated delivery method, in which SOD was encapsulated in biodegradable nanoparticles, making SOD-NP. This method yielded sustained protection against ischemia-reperfusion injury in rats by reducing edema, reducing ROS production and maintaining BBB integrity [66].
Despite the large number of animal studies showing protective effect of antioxidant strategy against cerebral ischemia-reperfusion injuries, there is still long way to go for clinical administration of this strategy. A clinical trial for patients with hemorrhagic shock demonstrated that 5 days of continuous intravenous injection of recombinant superoxide dismutase caused significantly improved neurological functional outcomes [67]. However, many human clinical trials of antioxidant therapy to prevent or attenuate ischemia-reperfusion injury have not yielded significant improvements [68]. Thus, the efficacy of antioxidant therapy in patients still needs to be validated, and further randomized human clinically trials are warranted.
Inhibition of leucocytes infiltration
Due to the critical implication of leucocytes infiltration in cerebral ischemia-reperfusion injury, strategies targeting leucocytes infiltration have great potential for developing effective treatment against ischemiareperfusion injury. These strategies include inhibition of leucocytes adhesion molecules synthesis, inflammation factor release and receptor-mediated leucocyte adhesion to endothelial cells. A big array of studies have confirmed the efficacy of this strategy in protecting against ischemia-reperfusion injury using inhibitors of leucocyte adhesion molecule synthesis, such as glucocorticoids and D-penicillamine [69]. For example, 10,17S-docosatriene, which is a DHA (Docosahexaenoic acid) messenger synthesized in DHA-oxygenation pathway, has shown potent inhibition for leucocytes infiltration and pro-inflammation gene induction, and has neuroprotection effect against ischemia-reperfusion injury [70]. This DHA messenger also inhibits cytokine-mediated proinflammation gene expression in cultured neurons. Another example is lipoxins that are potent inhibitors of leucocyte chemotaxis, adhesion and transmigration induced by inflammation factors, suggesting that they are part of innate protective pathways dampening the host inflammatory response. Chiang et al. [71] found that administration of biostable lipoxin analogues attenuated PMN-mediated vascular barrier dysfunction and second-organ injury in several models of ischemia/reperfusion.
Moreover, curcumin is one of the curcuminoids found in dried turmeric rhizomes with anti-inflammation property. Treatment with curcumin after MCAO but prior to reperfusion decreased neutrophil rolling and adhesion to the cerebrovascular endothelium, and eventually reduced brain infarct and edema, and improved neurological function outcomes [72]. Furthermore, two animal studies indicated that leukocyte depletion using anti-neutrophil monoclonal antibody treatment leads to better recovery of regional CBF and significantly reduces infarct size after cerebral reperfusion [35,36]. Similarly, anti- ICAM-1 monoclonal antibodies were also shown to reduce neutrophil infiltration and brain lesion size after ischemia-reperfusion in rats, while this effect was not seen in permanent ischemia [73]. Overall, these data suggest that neutrophil or ICAM-1 may be attractive targets for therapeutics development against ischemia-reperfusion injury. More experimental studies are required to further optimize these strategies, and eventually proceed to clinical translation of these strategies.
Platelet depletion or inhibition of platelet aggregation
Platelet is also a potential target for therapeutics against ischemiareperfusion injury. Previous studies have shown beneficial effect of platelet depletion in ischemia-reperfusion injury. For example, it has been demonstrated that platelet depletion using filter can improve both liver [74] and pancreas [75] function after ischemia-reperfusion injury, probably through reducing lipids peroxidation in cell membrane and the rate of thromboxane A2 prostaglandin I2. In addition, antiplatelet agents including dipyridamole and cilostazol have also been shown to improve myocardial function when combined with statin after ischemia-reperfusion [76]. These findings suggest that platelet depletion might be a potential therapeutic strategy for ischemiareperfusion injury in the brain as well.
Strategies targeting platelet aggregation may also benefit outcomes after ischemia-reperfusion. It was reported that an inhibitor of glycoprotein IIb/IIIa receptor, GPI 562, was able to inhibit GP IIb/IIIa receptormediated platelet aggregation and improve neurological outcomes after transient MCAO-reperfusion [77]. Additionally, as we mentioned earlier, thromboxane A2 (TXA2) is able to induce platelet aggregation and subsequent thrombosis, thus could be another therapeutic target. A recent study using this strategy in rat MCAO-reperfusion model showed that N2 (4-(2-(1H-imidazol-1-yl) ethoxy)-3-methoxybenzoic acid), an analog of Dazoxiben that has antiplatelet aggregative activity, was protective against MCAO-reperfusion injury through inhibiting TXA2 formation, platelet aggregation and thrombosis [78]. This antiplatelet strategy for treatment of ischemia-reperfusion injury still awaits further validation and the potential risk of bleeding caused by antiplatelet agents should be always taken into account.
Complement therapy
Experimental studies have demonstrated that inhibition of complement activation can protect against ischemia-reperfusion injury in various experimental models [57,79]. For example, C3 convertase is a member of serine protease family in the complement system. It has been found that an inhibitor of C3 convertase, the soluble complement receptor 1 (sCR1), significantly decreased myocardial infarct size and improved myocardial function in a rat model of ischemia-reperfusion [80]. More interestingly, the CR1 short consensus repeats has been confirmed to protect against cerebral ischemia-reperfusion injury in rats, decreasing cerebral infarct size and improving neurological function [81]. Additionally, C5 is another important component of complement system, which after cleavage can be converted into C5a and C5b-9, two potent inflammation mediators that increase vascular permeability, leucocyte adhesion and activation, and endothelial activation [82]. In a clinical investigation on patients undergoing coronary artery bypass grafting (CABG) surgery, the administration of a recombinant antibody against human C5, namely pexelizumab (Alexion Pharmaceuticals, Inc., Cheshire, CT), significantly attenuated complement activation, leucocyte activation, myocardial injury and acute postoperative mortality of these patients [83]. Moreover, C5a receptor antagonists were also shown to attenuate ischemia-reperfusion injury in animal models of stroke [79]. These findings strongly imply the potential therapeutic effect of complement inhibition strategies against ischemia-reperfusion injury. However, to date no many complement inhibitors are powerful enough to advance to human clinical trials. More potent complement inhibitory reagents targeting ischemia-reperfusion are yet to be developed in the future.
Post-conditioning
Post-conditioning emerges as another potential effective strategy for treatment of ischemia-reperfusion injury [84]. This strategy was first approved to be protective in the heart by intermittent reocclusion of the artery that perfuses the ischemic vascular bed [85]. Later on this strategy was applied in a rat stroke model with permanent MCAO plus transient bilateral common carotid artery (CCA) occlusion. After CCA reperfusion for 30 seconds, post-conditioning was performed by occluding CCAs for 10 seconds, followed by another two cycles of 30 seconds of reperfusion and 10 seconds of CCA occlusion. This treatment significantly reduced infarct size, probably by blocking apoptosis and free radical generation [86]. More studies further validated the protection effect and also investigated the mechanisms of post-conditioning in cerebral ischemia-reperfusion injury. For example, ischemia post-conditioning was shown to protect against 60 min MCAO and reperfusion, probably through reducing inflammation [87]. Furthermore, in another study remote post-conditioning was performed in rats after 90 minutes MCAO by occluding and releasing the femoral artery in the bilateral lower limb for three cycles, each occlusion and release lasting for 10 minutes. This treatment significantly reduced brain infarct volume, brain edema and BBB leakage compared to control groups [88]. The effect of post-conditioning might be time sensitive as Pignataro et al. showed post-conditioning was protective when it was implemented within 30 minutes of reperfusion, but not later [89]. More experimental investigations are warranted to further optimize the timing and dosing of post-conditioning before this strategy can be advanced to clinical application.
In summary, over the past few years there have been significant advancement in our understanding of molecular and cellular mechanisms of ischemia-reperfusion injury in the brain. The major mechanisms of reperfusion injury include oxidative stress, leukocyte infiltration, platelet activation and aggregation, complement activation and breakdown of BBB, which ultimately lead to edema or hemorrhagic transformation. A number of therapeutics studies are ongoing targeting these injury mechanisms, which however are still far from being clinically successful. Further investigations on the mechanisms of reperfusion injury are warranted, which will be helpful for developing effective therapeutics for ischemia-reperfusion brain injury.
This work was supported in part by NIH Grant R01-NS049476 (to X. Wang) and American Heart Association Scientist Development Grant (15SDG25550035) (to Z. Yu).