Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2013) Volume 0, Issue 0
Risperidone is one of the most widely used atypical antipsychotic drugs and is approved for the treatment of mental disorders (eg. schizophrenia, autism) in children and adolescents. The present study investigated the repeated treatment effect of risperidone and associated neurotropic mechanism in the phencyclidine (PCP)-induced hyperlocomotion model in adolescent rats. We examined whether repeated risperidone treatment would cause a sensitized inhibition of PCP-induced hyperlocomotion in adolescent rats, and whether such a sensitization effect was mediated by risperidone-induced alterations in Brain-Derived Neurotrophic Factor (BDNF), an important biomarker which plays a role in neuropathology of schizophrenia and action of antipsychotic medications. Male adolescent Sprague-Dawley rats (postnatal days [P] 44-48) were first treated with risperidone (0.5 or 1.0 mg/kg, sc) or vehicle and tested in the PCP (3.2 mg/kg, sc)-induced hyperlocomotion model for 5 consecutive days. Three days later, all rats were then challenged with risperidone (0.5 mg/kg) and PCP on ~P 51 to assess the potential sensitization effect. They were then sacrificed 1 day later and BDNF levels in the prefrontal cortex (PFC), striatum and hippocampus were examined using Western blotting. Behaviorally, repeated risperidone treatment progressively increased its inhibition of PCP-induced hyperlocomotion across the 5 test days. In the subsequent challenge test, rats previously treated with risperidone 1.0 mg/kg showed a stronger inhibition of the PCP-induced hyperlocomotion than those previously treated with vehicle, indicating a robust risperidone sensitization. However, no such group differences in the BDNF and its precursor proteins in any of the three brain regions were found. Therefore, although repeated adolescent risperidone administration induced a sensitization effect in the PCP-induced hyperlocomotion model in a dose-dependent fashion, whether BDNF is critically involved in this effect is still unsettled. Future work that directly manipulates BDNF systems is needed to further investigate this issue.
<Keywords: Brain derived neurotrophic factor; Risperidone; Adolescence drug treatment; Phencyclidine
Brain-derived neurotrophic factor (BDNF) is a member of the neurotrophin family that is widely distributed in the brain with the highest levels found in the hippocampus and other cortical areas [1]. It is involved in many functional processes critical for the brain development and experience-dependent neuroplasticity, such as neuronal survival, neural migration, differentiation, synapse formation, and modulation of neurotransmitter synthesis [2-4]. It also plays an important role in stress response, learning and memory, and actions of psychoactive drugs [5-7]. Accumulating clinical evidence indicates that abnormal BDNF expressions may contribute to pathophysiology of schizophrenia, as the levels of BDNF have been found decreased in peripheral (blood) and central (brain) systems of patients with schizophrenia [8-11]. Antipsychotic drugs can also alter the brain levels of BDNF, and prevent the stress-induced decrease in the levels of BDNF, indicating that it may also regulate the action of antipsychotic drugs [12-17].
In recent years, we have used the Conditioned Avoidance Response (CAR) and phencyclidine (PCP)-induced hyperlocomotion to examine the long-term treatment effects of antipsychotic drugs [18-23]. Both tests are known for their high predictive validity for antipsychotic efficacy, as antipsychotic drugs show a robust suppression of avoidance response and PCP-induced hyperlocomotion upon acute drug administration [24,25]. Testing an antipsychotic drug in two independent tests of antipsychotic activity is necessary to ensure that any observed antipsychotic effect is not an artifact of any particular model but reflects the generality of the treatment effect. We have been particularly interested in how repeated antipsychotic treatment alters this suppression over time. Such alterations can be manifested as either tolerance, which is characterized by decreased responsiveness to a certain drug effect, or sensitization, which is characterized by increased responsiveness to a drug effect [26]. The typical paradigm that we developed to study antipsychotic sensitization and tolerance consists of an induction phase, in which rats are repeatedly treated with an antipsychotic drug or vehicle for several days and tested for their avoidance responses and PCP-induced hyperlocomotion, and an expression phase, in which all rats are given a challenge dose of the drug and their avoidance and motor activity under PCP is tested again [18,19,21,22,27]. Using such a paradigm, we show that repeated treatment of haloperidol or olanzapine progressively increases its suppression of avoidance responding across the test sessions in the induction phase, and makes animals more sensitive to these drugs in the expression phase, evidenced by the findings that animals who have been treated with haloperidol or olanzapine in the induction phase show a significantly lower avoidance or lower PCP-induced hyperlocomotion than those treated with vehicle. In contrast, repeated treatment of clozapine tends to lose its avoidance suppression ability over time, thus causing a tolerance effect [18,27]. Risperidone (a widely used atypical antipsychotic drug) also causes a sensitization effect in the CAR test [23,28,29], but whether it would cause a similar effect in the PCP-hyperlocomotion test has not been examined. This is an important issue as it addresses the generality of repeated risperidone treatment induced sensitization phenomenon.
Despite the strong demonstration of risperidone sensitization in the avoidance conditioning task, the neurobiological mechanisms underpinning such an effect remain elusive. Because risperidone sensitization likely reflects a consequence of drug-induced brain changes, and BDNF is one important molecule involved in antipsychotic drug-induced neuroplasticity, we thus speculated that risperidone sensitization may depend on the drug-induced alteration of BDNF in the schizophrenia-related neural circuitry (e.g. the prefrontal cortex, hippocampus and striatum). In the present study, we tested this hypothesis by examining the risperidone-induced BDNF protein in adolescent rats (~P 52). We chose adolescent rats because our recent work shows that olanzapine or clozapine exposure can induce longterm alterations in responsiveness to antipsychotic treatment during adolescence in the CAR model [30]. It was of interest to explore whether risperidone could do the same. The choice of adolescent rats was also clinically significant as there has been a significant increase in the number of children and adolescents who are being treated with antipsychotic drugs in recent decades [31], and we do not know much about the long-term consequences of such early drug exposure.
Animals
Male Sprague-Dawley adolescent rats (P 33-37, 101-125 g on the delivery date, average age=~P 35) from Charles River Inc. (Portage, MI) were used. They were housed two per cage, in 48.3 cm×26.7 cm×20.3 cm transparent polycarbonate cages under 12-h light/dark conditions (light on between 6:30 am and 6:30 pm). Room temperature was maintained at 22 ± 1°C with a relative humidity of 45-60%. Food and water was available ad libitum. Animals were allowed at least 5 days of habituation to the animal facility before being used in experiments. All procedures were approved by the Institutional Animal Care and Use Committee at the University of Nebraska-Lincoln.
Drugs and choice of dose
The injection solution of phencyclidine hydrochloride (PCP, gift from National Institute on Drug Abuse Chemical Synthesis and Drug Supply Program) was obtained by mixing the drug with 0.9% saline. The dose of PCP (3.2 mg/kg, sc) was chosen based on our previous work [18,22,24,32]. This dose of PCP is shown to induce a robust hyperlocomotion effect without causing severe stereotypy [33,34]. Risperidone (RIS) (gift from the NIMH drug supply program) was dissolved in distilled sterile water with 1.0% glacial acetic acid. Two doses (0.5 and 1.0 mg/kg) were tested as they have previously been shown to acutely inhibit PCP-induced hyperlocomotion [24]. Furthermore, both drugs at these doses give rise to a clinically comparable range (60%-80%) of striatal dopamine D2 occupancy in rats, which is similar to values observed in schizophrenic patients [35]. All drugs were administrated subcutaneously (sc) at 1.0 ml/kg.
The motor activity testing apparatus is described in detail elsewhere [22,24,36]. Sixteen activity boxes were housed in a quiet room. The boxes were 48.3 cm×26.7 cm×20.3 cm transparent polycarbonate cages, which were similar to the home cages but were each equipped with a row of 6 photocell beams (7.8 cm between two adjacent photo beams) placed 3.2 cm above the floor of the cage. A computer detected the disruption of the photocell beams and recorded the number of beam breaks. All experiments were run during the light cycle.
Experimental procedure
Thirty-two adolescent rats (~P 42-43) were first handled, injected with 1.0% glacial acetic acid sterile water, and placed in the locomotor activity apparatus for 2 days (30 min/day), so they habituated to the experimenter’s handling and testing apparatus. They were then randomly assigned to 1 of 4 groups (n=8 /group): VEH+VEH (1.0% glacial acetic acid sterile water+saline), VEH+PCP (1.0% glacial acetic acid sterile water+PCP 3.2 mg/kg, sc), RIS 0.5+PCP (RIS 0.5 mg/ kg+PCP 3.2 mg/kg), and RIS 1.0+PCP (RIS 1.0 mg/kg+PCP 3.2 mg/kg) and were repeatedly injected with the drugs and tested for locomotors activity daily for 5 consecutive days. On each test day, they were first injected with vehicle, RIS 0.5 or 1.0 mg/kg, and then immediately placed in the boxes for 30 min. At the end of the 30-min period, they were taken out and injected with PCP (3.2 mg/kg, sc) or saline and placed back in the boxes for another 60 min. Motor activity was recorded in 5 min intervals throughout the entire 90-min testing session.
Two days after the last RIS test (~P 50), all rats were returned to the locomotor activity boxes for one re-habituation session (30 min), followed by the RIS challenge test 1 day later (~P 51). On the challenge day, all rats were first injected with RIS 0.5 mg/kg and then immediately placed in the motor activity boxes for 30 minutes. At the end of the 30-min period, rats were taken out and injected with PCP (3.2 mg/kg, sc) and placed back in the boxes for another 60 min. Motor activity was recorded for the entire 90-min testing session. One day after the challenge test (~P 52), rats were sacrificed by live decapitation and their brains were removed for further analysis [37].
Preparation of protein extracts
Rat brains were quickly removed and the prefrontal cortex (PFC), striatum and hippocampus were dissected out over ice according to the brain atlas of Watson and Paxions (5th edition) [38], and were frozen on dry ice and stored at -80°C for further analysis. Tissues from these areas were homogenized in ice-cold RIPA buffer, containing 25 mM Tris/HCl (pH 7.6), 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS (Thermo Scientific, Rockford, IL) with Protease Inhibitor Cocktail (Thermo Scientific, Rockford, IL). After centrifugation at 16,000 g for 15 min, the supernatant was collected and protein concentration was determined using the BCA Protein Assay Kit (Pierce, Rockford, IL).
Western blot analysis
Equal amount of proteins (40 μg/lane) was run on a sodium dodecyl sulfate (SDS)-12% polyacrilamide gel (Bio-Rad, Hercules, CA). Proteins were separated by electrophoresis at 80-120 V for 90 min (BDNF and β-actin), then electrophoretically transferred onto Polyvinylidene Fluoride (PVDF) membranes (Millipore, Billerica, MA) for 60 min at 300 mA in Tris/glycine buffer in a tank transfer system (Bio-Rad, Hercules, CA). The PVDF membranes were blocked with 5% nonfat dry milk in Tris-buffered saline (TBS) for 2 h at room temperature, and then incubated with primary antibody overnight at 4C. Immunostaining was carried out using the following antibodies. For BDNF, the membranes were incubated with a 1:100 dilution of anti-BDNF polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA), which recognizes both the mature form of the BDNF (14 kDa) and its precursor (pro-BDNF, 32 kDa). For β-actin (used as an internal standard), the membranes were incubated with anti-β-actin polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) with a 1:800 dilution. After washed 3 times in TBS with 0.1% Tween 20 (TBST) at a 10-min interval, the membranes were incubated with Odyssey anti-rabbit and anti-goat secondary antibodies (Li-COR Biosciences, Lincoln, NE), respectively, with 1:5000 dilutions in TBST at room temperature. After 1 h, the membranes were washed 3 times at a 10-min interval. Then the bands were visualized and quantified using Odyssey Fc Imager (Li-COR Biosciences, Lincoln, NE) according to the manufacturer’s instruction.
Four rats (1 each from the VEH+VEH and RIS 1.0+PCP groups, and 2 from the VEH+PCP) were identified as outliers based on the motor activity in the repeated drug test phase by the IBM®SPSS v19 program, and their data were excluded from subsequent data analysis. Motor activity data from the 5 drug test days were analyzed using repeated measures ANOVA (the between-subjects factor:drug group; the within-subjects factor:test day), followed by post hoc LSD tests to examine group difference. Differences between Day 1 and Day 5 during the adolescent drug test phase in individual groups were analyzed using a two-way (day×block) repeated measures ANOVA. Group differences on the challenge test were examined using a one-way ANOVA followed by post hoc Tukey tests. The BDNF protein was quantified by normalizing to β-actin which was re-probed on the same membrane and then calculated as percentage of the corresponding control group (deemed to be 100%). Group and regional differences on the BDNF protein were analyzed using repeated measures ANOVA [4 (group)×3 (region)], followed by post hoc Tukey tests.
Motor activity data
Five repeated drug test days: Figure 1A shows the mean motor activity of the four groups of rats during the 30-min test period before PCP or vehicle injection throughout the 5 days of drug testing. Twoway repeated measures ANOVA revealed a main effect of group, F(3, 24)=10.295, p<0.001, and a main effect of test day, F(4, 96)=36.828, p<0.001, and a significant group×day interaction, F(12, 96)=2.811, p=0.002. Post hoc Tukey tests revealed that the two RIS groups had significantly lower motor activity than the VEH+VEH group, all p<0.003, suggesting that RIS had a suppression of spontaneous motor activity. Interestingly, the VEH+PCP group also had significantly lower motor activity in this 30-min period before PCP injection than the VEH+VEH group, p=0.020, indicating that rats in the PCP withdrawal state exhibited a decreased motor activity which may resemble negative symptoms of schizophrenia.
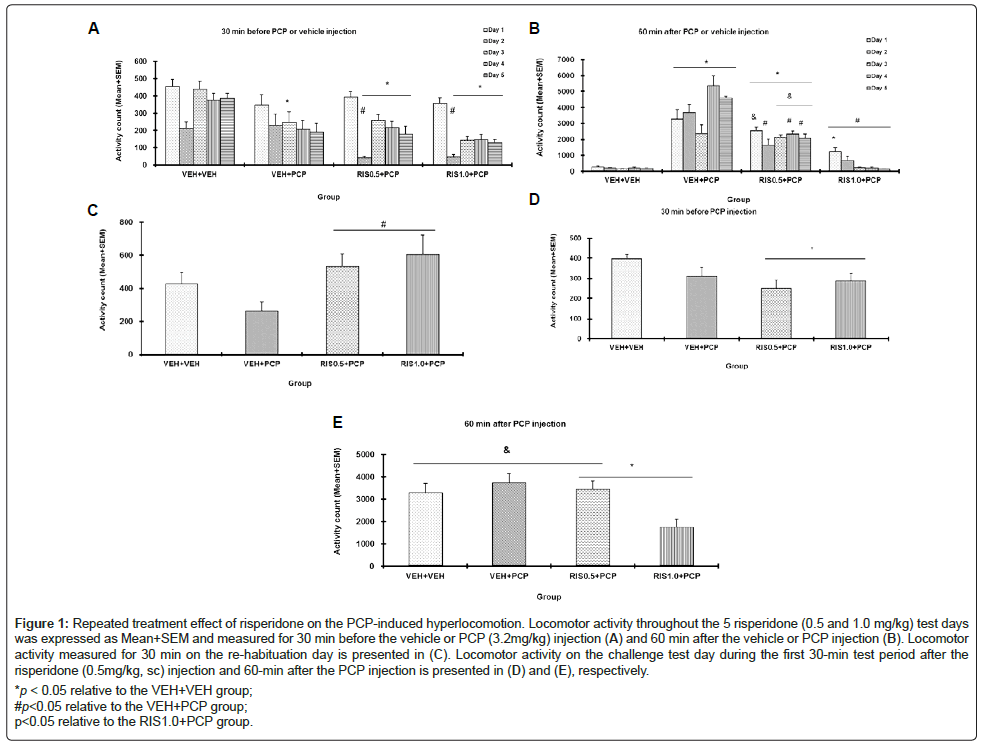
Figure 1: Repeated treatment effect of risperidone on the PCP-induced hyperlocomotion. Locomotor activity throughout the 5 risperidone (0.5 and 1.0 mg/kg) test days was expressed as Mean+SEM and measured for 30 min before the vehicle or PCP (3.2mg/kg) injection (A) and 60 min after the vehicle or PCP injection (B). Locomotor activity measured for 30 min on the re-habituation day is presented in (C). Locomotor activity on the challenge test day during the first 30-min test period after the risperidone (0.5mg/kg, sc) injection and 60-min after the PCP injection is presented in (D) and (E), respectively.
*p < 0.05 relative to the VEH+VEH group;
#p<0.05 relative to the VEH+PCP group;
p<0.05 relative to the RIS1.0+PCP group.
Figure 1B shows the mean motor activity of the four groups of rats during the 60-min test period after PCP or vehicle injection throughout the 5 days of drug testing. Two-way repeated measures ANOVA revealed a main effect of group, F(3, 24)=123.664, p<0.001, and a main effect of test day, F(4, 96)=5.508, p<0.001, and a significant group×day interaction, F(12, 96)=5.738, p<0.001. Post hoc Tukey tests revealed that the VEH+PCP group had significantly higher motor activity than the VEH+VEH group, p<0.001, indicating a strong psychomotor activation effect of this dose of PCP. The two RIS groups had significantly lower motor activity than the VEH+PCP group, all p<0.001, confirming the inhibitory effect of RIS treatment on PCP-induced hyperlocomotion. The inhibition by RIS 1.0 mg/kg was rather strong, as the RIS 1.0+PCP group had a comparable level of motor activity to that of the VEH+VEH group, p=0.499.
Re-habituation session
On the re-habituation day, all rats were placed in the motor activity boxes for 30 min with no drug treatment (Figure 1C). Rats previously treated with RIS showed much higher motor activity than the other groups. One-way ANOVA showed the group effect was marginally significant, F(3, 24)=2.973, p=0.052. Two group comparisons found that the two RIS groups differed significantly from the VEH+PCP group, p<0.031, suggesting a compensatory rebound effect against the motor inhibitory effect of RIS during the drug withdrawal.
Challenge test (sensitization assessment)
On the RIS challenge test, all rats were injected with RIS 0.5 mg/kg first, 30 min later, followed by PCP 3.2 mg/kg. In the first 30 min before the PCP injection, the two RIS groups had lower motor activity than the VEH+VEH group. One-way ANOVA showed the group effect was marginally significant, F(3, 24)=2.922, p=0.055.
In the 60 min after the PCP injection, the group difference was significant, F(3, 24)=4.922, p=0.008. Post hoc Tukey tests indicated that the RIS 1.0+PCP group had significantly lower motor activity than the VEH+PCP group, p=0.012, and significantly lower motor activity than the RIS 0.5+PCP group, p=0.022, suggesting that prior treatment of risperidone dose-dependently enhanced animals’ sensitivity to this drug in inhibiting PCP-induced hyperlocomotion, a clear sign of risperidone sensitization (Figure 1).
BDNF Data
As shown in figure 2, there did not appear to be any group and regional differences on the BDNF protein levels. Two-way repeated measures ANOVA with the group as the between-subjects factor and brain site (eg. PFC, hippocampus and striatum) as the within-subjects factors did not find a main effect of group, F(3, 24)=0.141, p=0.934, a main effect of brain site, F(2, 48)=0.463, p=0.632, or their interaction, F(6, 48)=0.338, p=0.913. Similarly, no significant group and regional differences on the Pro-BDNF proteins were observed. Two-way repeated measures ANOVA failed to find a main effect of group, F(3, 24)=0.141, p=0.934, a main effect of brain site, F(2, 48)=0.373, p=0.691, or their interaction, F(6, 48)=0.106, p=0.995 (Figures 2 and 3).
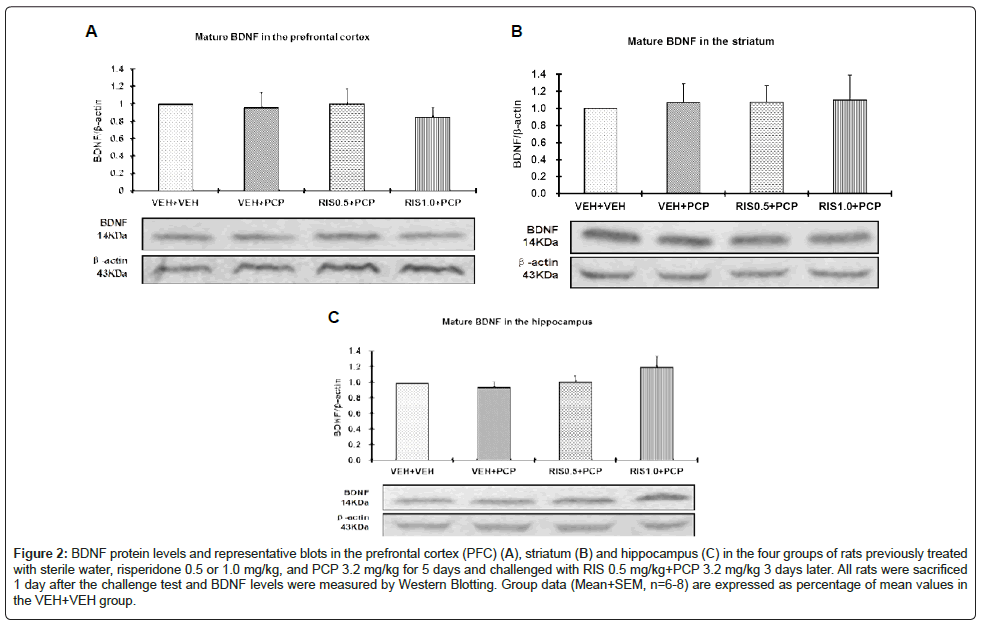
Figure 2: BDNF protein levels and representative blots in the prefrontal cortex (PFC) (A), striatum (B) and hippocampus (C) in the four groups of rats previously treated with sterile water, striatum-hippocampus 0.5 or 1.0 mg/kg, and PCP 3.2 mg/kg for 5 days and challenged with RIS 0.5 mg/kg+PCP 3.2 mg/kg 3 days later. All rats were sacrificed 1 day after the challenge test and BDNF levels were measured by Western Blotting. Group data (Mean+SEM, n=6-8) are expressed as percentage of mean values in the VEH+VEH group.
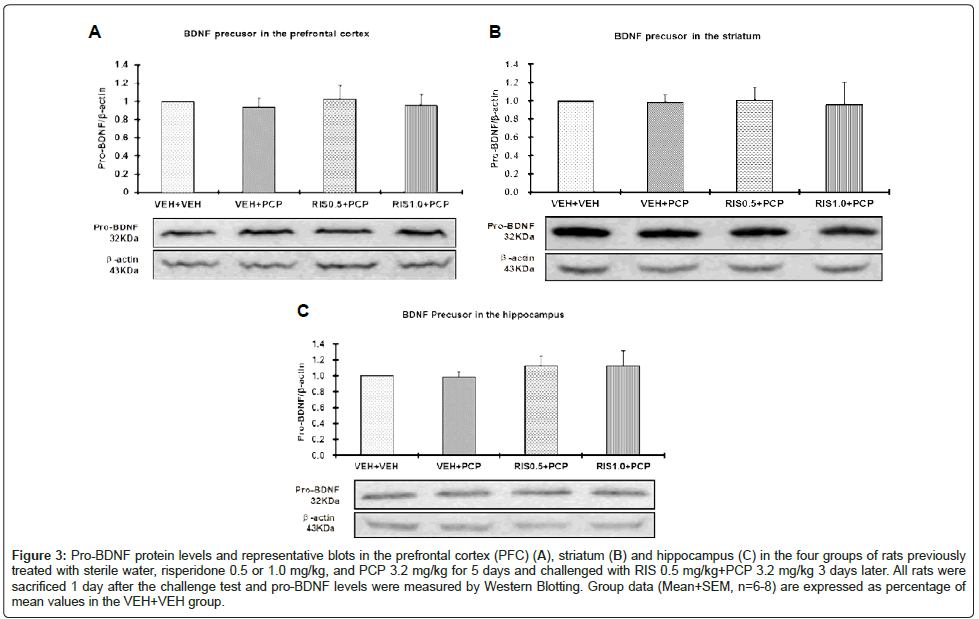
Figure 3: Pro-BDNF protein levels and representative blots in the prefrontal cortex (PFC) (A), striatum (B) and hippocampus (C) in the four groups of rats previously treated with sterile water, risperidone 0.5 or 1.0 mg/kg, and PCP 3.2 mg/kg for 5 days and challenged with RIS 0.5 mg/kg+PCP 3.2 mg/kg 3 days later. All rats were sacrificed 1 day after the challenge test and pro-BDNF levels were measured by Western Blotting. Group data (Mean+SEM, n=6-8) are expressed as percentage of mean values in the VEH+VEH group.
This study examined the long-term behavioral effects of repeated risperidone treatment in adolescent rats in the PCP-induced hyperlocomotion model, a well-known test for antipsychotic activity. Using a two-phase paradigm (i.e. an induction phase and an expression phase), we demonstrated that repeated administration of risperidone dose-dependently increased its inhibition of the PCP-induced hyperlocomotion across the 5 drug test sessions. In the challenge test, under the influence of risperidone 0.5 mg/kg, the previously risperidone-treated group (RIS 1.0+PCP) still exhibited a significantly higher inhibition of the PCP-induced hyperlocomotion than the drug naïve group (VEH+PCP). Both findings demonstrated that for the first time, a sensitization effect could be induced by repeated risperidone treatment in adolescent rats in the PCP-induced hyperlocomotion model. However, BDNF protein levels in the PFC, striatum and hippocampus did not show any significant group differences, a finding not paralleled to the behavioral results. Therefore, it appears that prior repeated risperidone treatment did not cause a significant change in the BDNF protein level that could support risperidone sensitization.
Previous work has shown that acute administration of many antipsychotic drugs inhibits the hyperlocomotor activity induced by acute administrations of PCP in adult animals [34,36,39]. Our recent work has focused on how repeated antipsychotic treatment affects the PCP-induced hyperlocomotion over time [18,22,24,32]. We and others show that in adult rats, repeated haloperidol, olanzapine or clozapine treatment, but not anxiolytic (eg. chlordiazepoxide) nor antidepressant treatment (eg. fluoxetine and citalopram) progressively potentiates their inhibition of PCP-induced hyperlocomotion across sessions and prolongs this action within sessions [24,32,40]. When the long-term effects are assessed in a subsequent challenge test, adult rats previously treated with olanzapine show an enhanced response to olanzapine (i.e. sensitization), while those previously treated with clozapine show a decreased response (i.e. tolerance) [18]. The present study extended this line of research into risperidone and showed that risperidone shares a similar behavioral profile with olanzapine but not with clozapine, possibly due to their similar receptor binding profiles against dopamine D2 receptors and 5-HT2 receptors [41]. Furthermore, this study extended our previous work into adolescent animals and showed that even adolescent rats exhibited risperidone-induced alterations in antipsychotic response. This new finding is in principle consistent with our recent study showing that olanzapine sensitization could be induced in adolescent rats in a conditioned avoidance response model [20], indicating the generality of antipsychotic sensitization independent of any particular behavioral model.
On the re-habituation day, we noticed that rats previously treated with risperidone were more active than those treated only with vehicle, indicating some kind of compensatory rebound when the inhibitory effect of risperidone was taken off. Risperidone has a strong antagonist action against dopamine D2 receptor and repeated administration could cause an increase of D2 receptor density in the prefrontal cortex, striatum (eg. caudate-putamen, nucleus accumbens), and hippocampus [42]. In adolescent rats, it has been reported that repeated treatment of risperidone (0.3, 1.0 and 3.0 mg/kg) for 3 weeks dose-dependently increased D2 receptor density in the medial prefrontal cortex and hippocampus [37]. The high dose of risperidone (3.0 mg/kg) also increased D2 receptors in the striatum [37]. This rebound in motor activity under drug-free may reflect antipsychotic withdrawal-induced behavioral hypersensitivity – a state of dopamine supersensitivity that is characterized by increased behavioral responses to the psychomotor stimulating effects of dopamine agonists such as amphetamine or apomorphine thought to result from hypersensitivity of dopamine D2 (especially D2 high) receptors [43-47]. A similar effect has been observed in human patients and is termed “neuroleptic-induced supersensitivity psychosis” [48,49]. Future work needs to determine the reliability of such an effect and its clinical implications.
BDNF is a member of neurotrophin family. Due to its important role in the regulation of the brain development and functions, and schizophrenia is thought to be a neurodevelopmental disorder, it attracts a fair amount of interest as etiological factor of schizophrenia and a potential therapeutic target [8,50,51]. A recent meta-analysis study examining blood BDNF levels in schizophrenia compared to age-matched healthy controls suggests that reduced blood BDNF levels in schizophrenia is a rather consistent finding [9]. Decreased BDNF levels in cortical areas and hippocampus in patients with schizophrenia has also been reported [52], consistent with findings from various animal models [8,12,50]. Human studies on the medication effects often report no significant effects of medications on the peripheral and central BNDF proteins, including risperidone [9,51,53]. However, animal work suggests that antipsychotics such as haloperidol, clozapine and high dose of risperidone (eg. 4.11 mg/kg) tend to decrease BDNF expression in the rat hippocampus [12,13,54] (but see [14]). Some even report that risperidone and haloperidol reduce BDNF in the frontal cortex [13]. Low dose of risperidone with acute and chronic treatment was without effect on the hippocampal BDNF [54]. Based on these findings, it is suggested that antipsychotic medications are unable to reverse the decreased BDNF levels in patients with schizophrenia [9] and the therapeutic properties of antipsychotic drugs are not mediated by stimulation of BDNF [12]. In the present study, we also did not observe any significant changes of BDNF protein in the prefrontal cortex, hippocampus and striatum that could be attributed to 5 days of repeated risperidone treatment, indicating that this neurotrophin is not likely to be an important molecule involved in risperidone sensitization.
The lack of risperidone effect on the brain BDNF could be due to several factors, of which several methodological issues are worth being discussed. The first one is that our subjects were adolescent rats who have different BDNF intrinsic levels and sensitivity to external stimulations than adult animals [55,56]. Therefore, risperidone may affect their brains differently than adult brains, an important issue worth pursuing. The second factor is that all our animals were challenged with risperidone (0.5 mg/kg) and PCP (3.2 mg/kg) before being sacrificed. The measured BDNF levels may have been altered by the acute exposure to risperidone and PCP, thus potentially masking any BDNF changes induced by prior risperidone treatment. Indeed, Lipska et al. [12] shows that acute clozapine and haloperidol decreased hippocampal BDNF levels in normal adult rats, but had either no effect or further lowered BDNF mRNA levels in the brains of rats with neonatal lesions of the ventral hippocampus, a validated animal model of schizophrenia. Our finding that repeated risperidone treatment did not alter BDNF protein levels in the prefrontal cortex, hippocampus and striatum in the rats repeatedly treated with PCP is consistent with this finding, and with the finding that repeated PCP treatment does not alter BDNF in adolescent brains [57]. Future work examining brain BDNF levels, as well as the density of BDNF receptors (TrkB, pTrkB, p75, truncated TkB) in these regions under the drug free condition is necessary to identify the true impact of prior antipsychotic treatment on this molecule and its receptors. Furthermore, to make sure that BDNF participates in the antipsychotic sensitization mechanisms, one can block the BDNF receptors with an antagonist (eg. K252a) or test antipsychotic drugs in BDNF knockout mice.
Taken together, the present study showed that adolescent repeated risperidone administration induced a sensitization effect in the PCPinduced hyperlocomotion model in a dose-dependent fashion. This sensitization effect is similar to the one observed in the conditioned avoidance response model in adult animals [28,58], suggesting that it might be an intrinsic effect associated with repeated risperidone treatment. The neurobiological mechanisms responsible for risperidone sensitization are still not clear. Further studies are needed to determine whether BDNF is critically involved in the risperidone sensitization.
All authors declare no conflict of interest.
This study was funded in part by the NIMH grant (R01MH085635) to Professor Ming Li.s.