Organic Chemistry: Current Research
Open Access
ISSN: 2161-0401
ISSN: 2161-0401
Editorial - (2012) Volume 1, Issue 3
Because of the growing need for more versatile tools in synthetic reactions as well as industrially facile chemical synthesis techniques for the mass production of polymers and pharmaceuticals, a novel approach to the cleavage and coupling of new C-C bonds is crucial. Electrochemistry is a sustainable means of performing a chemical reaction at room temperature without excessive heating; it also offers the possibility of using inexpensive and environmentally benign cross coupling catalysts focused on iron complexes as the organometallic electron-transfer mediator. Several examples are indicated throughout this editorial to give an understanding of the applications of voltammetric techniques as synthetic tools. The role of mandatory high-energy organic methods such as reflux and week-long photolysis can be replaced by sustainable aspects of electrochemical operations using the electron as a proven reagent in synthetic chemistry
The scope of many organic lab techniques involves tedious and expensive routines, such as photolysis, refluxing for hours, and the use of organometallic catalysts that are made with Ru [1], Pt [2], Mo [3], Rh[4], and Pd [5-7]. A recent major contribution to organic synthesis is olefin metathesis by two main research groups of Professors Grubbs and Schrock [1-3]. Their means of cross coupling using ruthenium and molybdenum complexes made the synthesis of new organic compounds more versatile. The 2010 Nobel Prize in Chemistry was given to Heck, Negishi, and Suzuki for their different palladium catalyzed crosscoupling reactions [5-7]. Despite prize-winning achievements, there are many aspects to improve with the aforementioned techniques.
The major problem with refluxing is the heat and energy consumed during the reaction. Most synthetic reactions have multiple steps where refluxing is a key technique; in fact, refluxing between nine and fortyeight hours is common place in order to prepare precursors for drugs and polymers [8,9]. The excessive energy input in heating a swirling flask for hours or days at a time can be expensive and wasteful given the low yields for final products. Even with the addition of a catalyst, the need for reflux to initiate the first step of the reaction is probable [10]. The catalyst will indeed lower the activation energy of the reaction; however, the significant energy barrier overcome by these catalysts comes with a large price.
Most catalysts utilize an expensive transition metal center, which makes mass production of these compounds impractical. For instance, the values of ruthenium, palladium, rhodium, and platinum, listed in order of increasing cost, are anywhere between $15 and $130 per gram [11]. Despite the impairing expense of these rare earth metals, the synthesis of the catalysts is costly as well. The general price range of Ru and Mo-containing catalysts is $60 to $300/0.1 gram [12]. In order to carry out the designed syntheses, (When it comes to carrying out the designed synthesis), the daunting expense of the catalysts mentioned is indisputable. The price of the rare earth metal as well as the organic steps taken to synthesize the catalysts drive the cost upward, making the compounds expensive endeavors in research and development for both industry and academia.
The toxicity of these metal catalysts is also a significant dilemma. The most important metals involved in many of the Grubbs and Schrock Catalysts for olefin metathesis are ruthenium, which is carcinogenic if exposed over a period of time, and molybdenum, which is toxic, causing hyperbilirubinemia and gout [13-15]. For the palladium catalyzed heterolytic C-C coupling reactions, the metal center is one of the more expensive rare earth metals [11,12], making olefin metathesis and cross coupling impractical compared to current electrochemical applications presented below. The cost for many C-C coupling reactions due to these catalysts comes with a great price: not only is the opportunity cost of time and low yields involved, but health and physiological impediments must be taken into consideration along with the exorbitant expense of the transition metal itself.
Regarding synthetic chemistry, the basic concept of electrochemistry is breaking and making bonds using the smallest chemical reagent known to man: the electron. Environmentally idle solvents and inert salts as supporting electrolyte systems are coupled with the use of relatively inexpensive and reusable electrodes, such as carbon (graphite), nickel, or silver to increase the overall efficiency and effectiveness of the reactions.
Gaining (reduction) or loss (oxidation) of an electron by an appropriate chemical redox reagent is characteristic of many organic reactions. For instance, a reaction of an alkyl group on an aromatic ring with at least one hydrogen on the carbon attached to the ring with KMnO4 will completely oxidize that position, producing a carboxylic acid [16]. On the other hand, reduction can occur on a palladium surface in the presence of hydrogen or by using LiAlH4 or NaBH4 as a strong reducing agent [16]. These well-known oxidations and reductions in organic synthesis are manifest; however, electrochemistry works in a similar manner without using chemical redox reagents that could be harmful. Caustic KMnO4 can cause gastrointestinal defects if accidentally ingested as well as skin defatting if used over an extended period of time [17]. An environmentally benign and effective tool for organic synthesis is thus needed to avoid the onset of harmful conditions or diseases caused by common redox reagents and catalysts in organic chemistry.
Varying voltammetric techniques allow analysis and understanding of the kinetics behind the mechanism for electron-transfer reactions. Some pulse voltammetries are even capable of detecting analyte in trace quantities (ppm–ppb range) [18]. The most popular of these various techniques, however, is cyclic voltammetry (CV), which utilizes a nonlinear concept of measuring current as a function of potential. Some functional groups in compounds are able to show a unique anodic oxidation or cathodic reduction potential, denoted half potential (E1/2), versus the extra-stable ferrocene/ferrocenium couple (Cp2Fe0/+) [18-21], which is used as an internal standard material within the potential window dependent on the reaction media. The voltammetric scanning in the specified potential range back and forth in CV can be beneficial in detecting electron-transfer reactions that are reversible. In fact, the most pertinent ramification of cyclic voltammetry in a short time period (>10 s) is its ability to detect chemical reversibility, which is important in judging whether or not an electrocatalyst can be rejuvenated after electrolysis.
The most apposite organic synthetic tool within electrochemistry, however, is controlled-potential coulometry to produce thermodynamically stable products. This technique considers the potential at which the electron-transfer mediator or the organic molecule is activated by the electrode and continuously applies that potential to create some product via a radicalization reaction mechanism [22]. An electrolysis which lasts for one hour can have the same effect as an extended photoloysis experiment, an example discussed in the next section. Breaching the activation energy barrier using an electron takes advantage of the intense reactivity of the free radical created during electrolysis, and known electroanalytical observations can account for the mechanistic aspects of these reactions [18-22].
Electrochemistry has the potential (no pun intended) to be a versatile tool in organic reactions if given the proper attention by chemists pursuing new heterolytic alkylation (C-C coupling) reactions. One may use electrochemistry to cleave and create bonds, make reaction conditions more favorable, or analyze a reaction’s electrontransfer kinetics. Perhaps the most attractive aspect of electrochemistry is the mild and environmentally friendly conditions under which the reactions proceed (vide infra).
Electrochemistry offers an energy-efficient means of running a reaction under environmentally mild conditions. Instead of refluxing for forty-eight hours using temperatures exceeding 100°C, bulk electrolysis in synthetic scale could be employed as a desired tool by which bonds are broken and new bonds are formed in less than an hour at room temperature [18-25]. Electrolysis would then lead to a radical reaction through which electrons coalesce to form new bonds and create new thermodynamically stable products.
Novel routes to the formation of new C-C bonds both heterolytically and homolytically are imperative in the growing need for new compounds to allay the effects of many diseases, such as neurodegenerative disorders, cancer, HIV/AIDS, etc., or work as an agonist/antagonist in various physiological systems. Many organic compounds have electrochemical reactivity. For instance, our lab has taken advantage of the electrochemical behavior of the [2-benzyloxy- 1-methylpyridinium][OTf] (OTf =-OSO2CF3) salt (BOMPT), which has a cathodic reduction peak observed at Epc = -1.6 V vs. Cp2Fe0/+ in acetonitrile solvent using 0.1M [NBu4][OTf] as a supporting electrolyte at room temperature [23-24]. This electrolysis uses the power equivalent of two AA batteries. When electrolysis proceeded at ca. Eappl = -1.7 V vs. Cp2Fe0/+ for 20 min, many products formed stoichiometrically thereafter: bibenzyl (major), benzaldehyde, pyridinone (major), and toluene [25]. In the presence of iodomethane, however, the reaction produced ethylbenzene, iodotoluene (major), and bibenzyl (major) [25]. The reaction cathodically induces the reactivity of a halogen-free benzylation reagent at room temperature, which possibly promotes heterolytic and homolytic C-C coupling reactions.
Electrochemical cleavage of C=C bonds has also been under investigation. Consider the electrolysis reaction using p-methoxybenzalmaloninitrile: when applied at ca. Eappl = -1.8 V vs. Cp2Fe0/+ for 40 min in acetonitrile with 0.1M [NBu4][PF6] as a supporting electrolyte, a hydrolysis reaction seemingly cleaves the C=C double bond as a follow-up chemical reaction, forming p-methoxybenzaldehyde and propanedinitrile (Eq. 1) as the major products [26,27]. The C=C and C≡C bond cleavage reactions use substrates such as Pd(OAc)2, ZnCl2·2H2O, and CF3CO2H at 100°C for nearly 24 hours while under acidic conditions [28] in order to break the multiple bonds. The method outlined in our research suggests an electrochemically induced olefinic cleavage reaction, bypassing the expensive and superfluous substrates.
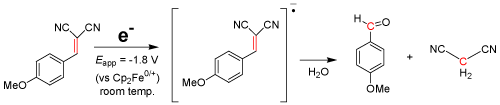 EQ. 1
EQ. 1
Given the nature and characteristics of the above equation, the conclusion is simple: Not only can electrochemistry be used to form new bonds, but the same method can be applied to breaking strong bonds, creating either useful materials or a process to render chemicals impotent, which is further discussed in the next section. Some chemicals, however, have no reduction or oxidation potential within the potential window of the supporting electrolyte and solvent system; therefore, an electrocatalyst must be introduced to initiate the reaction.
Electrocatalysis is a tool of exploiting the redox capabilities of organometallic complexes. The electrocatalytic behaviors of some organometallic compounds are well understood in the literature [17- 25]. These behaviors depend on the ability of the complex to accept or expel an electron. Chong and Geiger et. al. report an extensive study on [Re(II)(η5−C5H5)(CO)3]+ as an electrocatalyst capable of [2+2] coupling interactions with cyclooctene, cycloheptene, cyclohexene, and cyclopentene (for each cycloolefin, ca. 80% isolated dimerized yield obtained post electrolysis) [28]. The [2+2] coupling of cyclooctene is shown in equation 2 below.
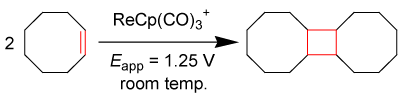 EQ. 2
EQ. 2
Story et. al. reported the equation above as the precursor to olefin metathesis reactions focused on the production of macrocyclic compounds [29], citing that there is little literature regarding the dimerization of other cycloolefinic compounds such as those previously mentioned. Many olefin coupling reactions yield between 2% and 80% product [29-31], where the best yields from C-C couplings and catalysts equally match the preliminary yields from the electrochemicallyinduced cyclization reaction of 80%; however, this electrochemical technique takes less time and is capable of utilizing benign and inexpensive catalysts, discussed below with diiron complexes. The rhenium compound undergoes anodic oxidation at a potential of Eappl >1.25 V vs. Cp2Fe0/+, meaning at this potential, the Organo-Re (II) electron-transfer mediator will give up an electron and seek an electron in solution for stabilization, most possibly from a nearby π-bond, taking advantage of weakly held olefinic π-electrons.
Recently, our lab has focused on the formation of C-C bonds via an anodically induced [2+2] cyclization reaction at room temperature using terminal alkenes (1-octene and 1-hexene), similar to the one that occurred to cyclooctene in the above equation. These [2+2] cyclization reactions would lay the groundwork for novel synthetic routes to overcome activation energy barriers in producing various cyclobutyl derivatives [32], which can be involved in different aspects of medicinal chemistry as well as industrial processes for macrocyclic polymerization. The equation given below represents preliminary research on the ability of this Organo-Re(II) electrocatalyst to induce cyclization reactions of terminal alkenes in creating syn- and anti-isomers of cyclobutyl derivatives following Woodward-Hoffman rules [33].
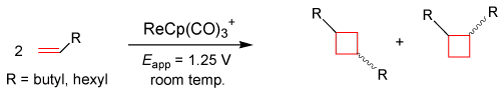 EQ. 3
EQ. 3
The Organo-Re(II) electron-transfer mediator shows reversibility in CV, which allows rejuvenation of the catalyst after electrolysis has taken place by running electrolysis where the new reducible species has formed in CV timescale. As mentioned before, the reaction shown above employs the reusable nature of the electrocatalyst in creating various cyclobutyl derivatives from terminal alkenes. Spee et. al. report seven-day photocatalysis dimerization products of [2+2] cycloadditions with yields up to 20% [34]. Preliminary results in our laboratory suggest yields as high as 70% after 1 hour of electrolysis, making electrochemistry under mild conditions a much more efficient tool than photocatalysis.
Heterolytic and homolytic C-C coupling reactions can take place via electrode-induced radical reaction mechanisms. The above compounds, such as BOMPT and p-methoxybenzalmalanonitrile, undergo reduction, which creates a radical anion possibly capable of various alkylations. If these reactions are studied further by the scientific community, the electrochemical method of electrolysis has the promise of providing a versatile tool for various coupling reactions.
The cleavage of C=C bonds in p-methoxybenzalmalanonitrile at room temperature is important because it allows a basis upon which the cleavage of the C=C bond of o-chlorobenzalmaloninitrile (CS/tear gas) can be introduced. The conversion of a harmful compound to one or more inert compounds (the hypothesized products for the reduction of tear gas are neutralized propanedinitrile and o-chlorobenzaldehyde) is necessary in the growing relevance of using such compounds as chemical or biological warfare. Rendering potent and harmful materials benign to physiological systems is easily and affordably employable through electrochemistry.
Catalysis can be a concrete tool used to decrease the activation energy of seemingly impossible reactions; however, as the introduction to current organic techniques suggests, there are many troubles with catalysts. Even as an electrocatalyst, rhenium is an expensive rare earth metal, which makes mass production of catalysts such as these expensive and impractical. Future studies into organometallic transition metal centers that are inexpensive and non-toxic are required with the increasing need for these catalysts in organic synthesis. One potential candidate is the diiron-carbonyl complex, which has recently shown behavior in CV timescale akin to the Organo-Re(II) electrocatalyst [31]. The research with diiron complexes is under intensive investigation and shows promise in C-C coupling catalysis.
A conclusion can be recognized from initial electrochemical data that similar cyclobutyl groups, such as those shown above produced by the Organo-Re(II) electrocatalyst, are possibly attainable using these iron-based dimers. Preliminary results in our lab using diiron carbonly complexes suggest the production of these cyclobutyl derivatives (yield ca. 20%) via anodically induced [2+2] couplings as well as linear addition products, as proposed by the following equation [35]:
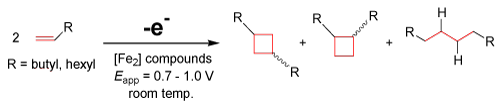 EQ. 4
EQ. 4
The reversible nature of these economically feasible diiron complexes to form new C-C bonds can be utilized. Reversibility is important because it takes advantage of the electrochemical ability of rejuvenating the catalyst after electrolysis. Iron is abundant, inexpensive, and friendly to ecosystems, which bypasses the costly and biologically destructive nature of the current and most commonly used transition metal catalysts. Iron-based complexes show promise as environmentally friendly candidates for the active site in many electrocatalysts. These electrocatalysts could potentially be capable of olefin metathesis [30,31] and various other coupling reactions. Taking advantage of the mild potentials of these electrocatalysts gives rise to a new method of C-C coupling reactions utilizing electrochemistry, where the versatility, sustainability, and overall efficiency of these reactions are much worth the research.
Electrochemistry as a synthetic tool may one day soon be conventional. The application of the electron as a powerful redox reagent could be analogous to using H+ in acid catalysis. The picking and choosing of solvent, electrolyte, electrode, and applied potential systems for an analyte and electrocatalyst could be as common as choosing reflux, photolysis, or excessive boiling and stirring to drive a reaction to completion. And perhaps most importantly, the scribbling of the number of electrons and, if needed, a necessary catalyst over a yield arrow shall be as routine as writing hν, reflux, or any common reagent currently used in all organic chemistry texts.
The introduction of organometallic catalysts to organic synthetic reactions allowed chemists a convenient means of new C-C couplings reactions [36]; however, the pursuit and exploration of electrocatalysis of more fiscally and environmentally practical catalysts is an issue which cannot be ignored. We must rise to the occasion in acknowledging society’s need for novel methods for creating new pharmaceuticals; innovative answers to questions of sustainability, and the unique opportunity to efficiently utilize the ever-so ubiquitous electricity soaring through our wires. Through electrochemical methods, organic chemists have a means of quick and efficient reactions, which provide higher yields than do current organic techniques, as well as environmentally sustainable procedures for inexpensive C-C coupling reactions. Because the potential prosperity of such a game-changing science is too persuading to bypass, embracing the possibility of being at the forefront of a scientific revolution is critical, and electrochemistry provides the enhanced versatile and sustainable necessities in a world where versatility and sustainability are paramount.
The authors would like to thank the following people and organizations for their efforts and financial contributions: Mr. Joshua May, Mr. Jinyu Liu, Miss My Bui, Ball State University ENHANCE Fund, and the Cardinals Fellowship. The authors extend a special thanks to Drs. Robert E. Sammelson, Philip A. Albiniak, and Jesse W. Tye for generous donations of malononitrile derivatives, the BOMPT compound, and diiron carbonyl complexes, respectively.