Enzyme Engineering
Open Access
ISSN: 2329-6674
ISSN: 2329-6674
Research Article - (2015) Volume 4, Issue 2
Diabetes mellitus is an important cause of mortality and morbidity worldwide [1]. Type 2 diabetes mellitus is characterized by inappropriate regulation of hepatic glucose production, which is mainly due to an imbalance in the relationship between glucagon and insulin levels in plasma. To maintain normal blood glucose levels during the fasting state by inducing hepatic glucose production is the major biological action of glucagon [2]. Glucagon exercises its action through glucagon receptor (GCGR) activation. GCGR activation leads to activation of signal transduction pathway, resulting in further activation of adenylate cyclase, which initiates cAMP production, in turn activation of the protein kinase A, finally leading to elevation of blood glucose levels [3]. These observations have developed interest in blockade of GCGR activity for the control of over production of liver glucose or the treatment of type 2 diabetes mellitus. GCGR belongs to class B (also known as secretin-like) family of G-protein coupled receptors (GPCRs) [4]. The GPCRs of this family are composed of an extracellular N-terminal domain and another domain of seven membrane-spanning α-helices [5]. Glucagon is a peptide hormone composed of 29 amino acids and is secreted by α cells of pancreas as a result of low blood glucose levels. Glucagon plays a significant role in glucose homeostasis [2]. There are many documented glucagon receptor antagonists which have been shown to efficiently terminate glucagon receptor action. Most of them either belong to the category of glucagon neutralizing antibodies [6] or are having low molecular weight [7-12]. There are various safety, tolerability and immunological concerns with these advances. These concerns are also, in part, our motivation behind this in-silico study to identify a favorable therapeutic choice (Figure 1).
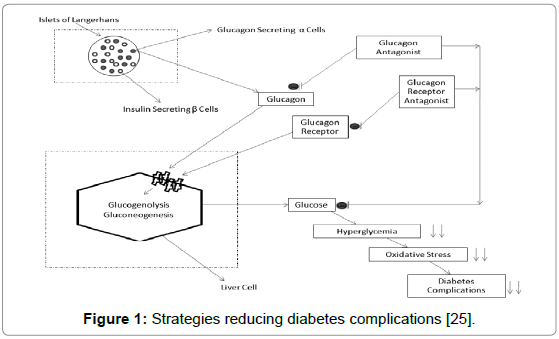
Figure 1: Strategies reducing diabetes complications [25].
Earlier it was difficult to discover glucagon receptor antagonists due to non-availability of its crystal structure. A GCGR-ligand binding model based on the approach of virtual screening study using modeled structure has been proposed [13]. However because of recent availability of the crystal structure of GCGR, now an in-silico rational drug design strategy can be applied to it [14]. In this study, using high throughput virtual screening strategy various compound libraries or databases containing thousands of compounds were screened against crystal structure of GCGR. Computational screening resulted into the discovery of a new GCGR antagonist which can obstruct the GCGRglucagon binding.
All computational studies were performed on a CentOS Linux operating system running on a HCL machine with an Intel Core i5 processor with 6 GB RAM.
Protein preparation
The crystal structure of human GCGR [PDB ID: 4L6R] at 3.40 Å resolution was retrieved from the Protein Data Bank [14]. The structural features, catalytic residues and active site residues of GCGR were analyzed by means of PDBsum [15]. For further studies, preparation of protein structure was processed through “Protein Preparation Wizard” of Maestro v9.7 interface of Schrödinger [16]. Protein preparation process involved assigning bond orders, addition of hydrogen bonds, creation of disulphide bonds, conversion of selenomethionine to methionine, filling of missing residues using Prime, capping of termini, deletion of waters and optimization. Energy was minimized using the OPLS_2005 molecular mechanics force field with default value of cut off RMSD (Root Mean Square Deviation).
Ligand library preparation
Thousands of compounds were extracted from the ZINC database ([https:// zinc.docking.org/browse/catalogs/natural-products) and InterBioScreen database (https://www.ibscreen.com) [17] and which are further processed with LigPrep v2.9 wizard of Maestro v9.7 interface of Schrödinger [18]. It involves generation of maximum possible isomeric and ionization variants. Applying Lipinski’s filter, the ligands having poor pharmacological properties were discarded to prepare a virtual library having pharmacologically preferred ligands.
Grid generation
As per the reported literature available, Tyr 138, Gln 142, Tyr 149, Val 191, Gln 232, Trp 295, Thr 296, Asn 298, Glu 362, Phe 365 and Leu 386 residues of GCGR are functional residues involved in glucagon binding [19]. A receptor grid was generated in the region of these residues of GCGR using Glide v6.2 of Maestro v9.7 interface of Schrödinger with default parameters.
Virtual screening and docking studies
A lead molecule with best docking score was retained through implementation of three subsequent docking operations such as HTVS, SP and XP using Glide v6.2 of Maestro v9.7 interface of Schrödinger [20]. Based on XP GScore, favorably docked ligands were ranked [21-23].
Comparison of the proposed lead molecule with some already reported GCGR antagonists
The structures of some already reported compounds with GCGR inhibitory activity were collected from different sources and after processing them using LigPrep v2.9 wizard of Maestro v9.7 interface of Schrödinger, these were also docked against same grid generated earlier. With reference to docking scores for proposed lead molecule and published GCGR antagonists, a comparative study was performed [24].
Molecular dynamics simulation
Molecular dynamics simulation was performed to evaluate the stability of proposed lead compound. It was performed using Desmond Molecular Dynamic System v4.0 [25] with OPLS_2005 force field [26]. First, the docking complex was processed through Desmond system builder wizard. The system was solvated in a box of SPC water having orthorhombic shape and minimized volume of 747168 Å3. Four chloride ions were added to neutralize the system. Default protocols were applied in minimization wizard of Desmond. MD simulation was run on this system for a time period of 5 nanoseconds. The temperature was fixed at 300 K temperature and pressure at 1.01325 Bar throughout the simulations process. After every 1.2 picoseconds and 4.8 picoseconds, frames were recorded to form energy representations and trajectory respectively. The model system was relaxed before simulation using default relaxation protocols. Finally, RMSD calculations were done for the entire simulation trajectory in reference to the first frame.
Prediction of pharmacokinetics in human body and comparison of the results with some already reported GCGR antagonists
To evaluate the pharmacokinetic properties QikProp v3.9 module of Maestro v9.7 interface of Schrödinger was used [24]. Various physico-chemical descriptors were calculated to further account for the potential of the lead molecule to act as efficient drug candidate. Violation of Lipinski’s rule, if any, was assessed using obtained values for these physico-chemical descriptors. With reference to these values for proposed lead molecule and published GCGR antagonists, a comparative study was performed.
Virtual screening and docking studies
Thousands of compounds extracted from ZINC and InterBioScreen databases were screened against the binding pocket of the prepared protein structure of Human GCGR. Three subsequent docking procedures such as HTVS, SP and XP were implemented using Glide v6.2 of Maestro v9.7 interface of Schrödinger [20,27]. Based on XP GScore, favorably docked ligands were ranked [21-23]. To find the top poses of the ligands, Glide E-model was used. The lead compound, STOCK1N82694 has a Glide Score of -9.70 and had good binding affinity for the GCGR receptor. Similar docking parameters for already reported GCGR antagonists are also reported in Table 1 for the comparative purpose. The docking studies indicated that the proposed lead compound STOCK1N82694 showed strong hydrogen bond and hydrophobic interactions with the important glucagon binding residues of GCGR. The lead compound STOCK1N82694 occupies the better binding efficiency against GCGR with higher docking score and strong interaction in comparison of some already published GCGR antagonists
| Compounds | Docking Score | Glide GScore | Glide e-model |
|---|---|---|---|
| BAY 27-9955 | -6.21 | -6.21 | -42.9 |
| 3-chloro-4-hydroxybenzoic acid (4-hydroxy-1-naphthyl-methylene)hydrazide | -4.83 | -7.24 | -32.8 |
| CP 99-711 | -4.96 | -4.96 | -33.8 |
| NNC 92-1687 | -2.78 | -5.22 | -37.0 |
| L-168,049 | -5.34 | -5.34 | -51.9 |
| NNC 25-0926 | -8.12 | -8.12 | -62.8 |
| Skyrin | -6.44 | -6.61 | -30.3 |
| Spirourea | -4.92 | -4.92 | -35.7 |
| Benzimidazole | -3.22 | -3.25 | -19.4 |
| STOCK 1N-82694 | -9.61 | -9.70 | -57.8 |
Table 1: Comparison of binding efficacy of some already published GCGR antagonists and STOCK 1N-82694 against GCGR.
Binding mode analysis of GCGR-STOCK1N82694 docking complex
The virtual screening result showed that STOCK1N82694 had the lowest docking score of -9.70 compared to already reported GCGR antagonists. The good binding affinity of STOCK1N82694 is due to various interactions with important glucagon binding residues of GCGR. Various interactions along with residues involved in interatomic contacts are reported in Table 2 and are also shown in Figure 2.
| GCGR-ligand | No. of H Bonds | Residues involved in H-ond formation | Other non-bonded interactions |
|---|---|---|---|
| STOCK1N82694 | 03 | Gln 232, Thr 296(2) | π-π cation (Trp 295, Phe 365); Hydrophobic (Tyr 149, Val 191, Met 231, Phe 303, Leu 307, Leu 382, Leu 386), Charged +ve (Lys 187, Arg 308, Arg 378, Lys 381), Charged -ve (Asp 385) and Polar (Gln 293, Ser 297, Ser 389) |
Table 2: Interactions of STOCK1N82694 with the active residues of GCGR.
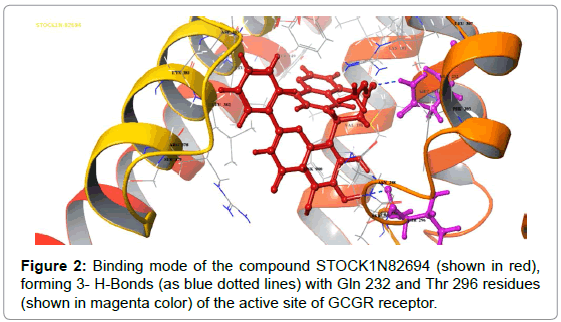
Figure 2: Binding mode of the compound STOCK1N82694 (shown in red), forming 3- H-Bonds (as blue dotted lines) with Gln 232 and Thr 296 residues (shown in magenta color) of the active site of GCGR receptor.
A deep cavity is formed by seven transmembrane helices in GCGR to provide binding region for ligand. Here, STOCK1N82694 interacted with GCGR through one hydrogen bond and a number of other contacts. Gln 232 and Thr 296 are the amino acid residues involved in the formation of hydrogen bonds with the ligand STOCK1N82694. Seven hydrophobic contacts with the amino acid residues Tyr 149, Val 191, Met 231, Phe 303, Leu 307, Leu 382, Leu 386 and four positive charge interactions with residues Lys 187, Arg 308, Arg 378, Lys 381 were observed. It also forms interactions with polar residues Gln 293, Ser 297 and Ser 389. The amino acid residues Trp 295 and Phe 365 are involved in the formation of π-π cationic interaction with the ligand STOCK1N82694. The ligand STOCK1N82694 also forms a -ve charged interaction with Asp 385 amino acid residue. The above mentioned interactions of STOCK1N82694 with various residues of GCGR propose it to be a potential ligand which could obstruct the glucagon- GCGR interaction.
Molecular dynamics simulation
Molecular dynamics simulation was performed to evaluate the stability of bound protein with proposed lead compound STOCK1N82694 (docking score=-9.61) [28]. There was performed an analysis of combined trajectory files for the flexibility of Cα atoms of GCGR-STOCK1N82694 complex. After proper alignment of all frames (1001), using Visual Molecular Dynamics (VMD) the RMSD values were plotted against the frames obtained during 5 ns simulation run. Initially, the RMSD values were found to be fluctuated between the minima and maxima of 0.971 Å and 2.754 Å respectively. (Average RMSD=1.886 Å). During 5 ns simulation time, the value of overall standard deviation for RMSD was observed to be 0.0237 Å (Figure 3).
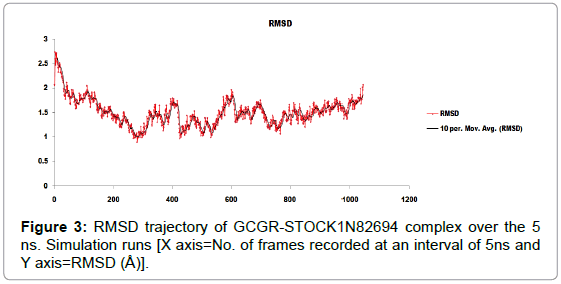
Figure 3: RMSD trajectory of GCGR-STOCK1N82694 complex over the 5 ns. Simulation runs [X axis=No. of frames recorded at an interval of 5ns and Y axis=RMSD (Å)].
During initial stage of 5 ns simulation run, potential energy was high but later on there was a decrease in the potential energy and after 1.5 ns of simulation, the potential energy variations was found to be in a fixed range which suggests the stability of the system (Figure 4).
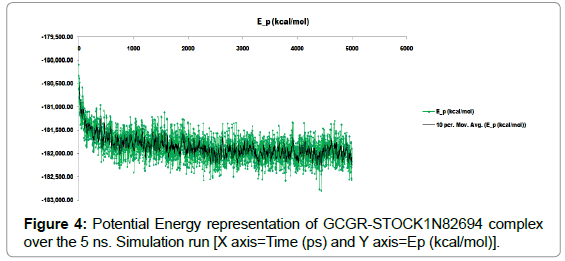
Figure 4: Potential Energy representation of GCGR-STOCK1N82694 complex over the 5 ns. Simulation run [X axis=Time (ps) and Y axis=Ep (kcal/mol)].
ADME properties prediction
Pharmacokinetic properties such as Molecular weight, Hydrogen bond donors, Hydrogen bond acceptors, log P (Octanol/water partition coefficient), percentage of human oral absorption, CNS activity and blood brain barrier partition coefficient are important for ADME estimation. All these values for STOCK1N82694 are following the recommended ranges (Table 3) for a drug with good pharmacological properties. This depicts the excellent potential of STOCK1N82694 as prospective lead to function as GCGR antagonist.
| Compound | MW | HBD | HBA | log P | % Oral Absorption | CNS Activity | BBB Partition Coefficient |
|---|---|---|---|---|---|---|---|
| BAY 27-9955 | 342.496 | 1.000 | 1.700 | 6.177 | 100.000 | 0 | -0.002 |
| 3-chloro-4-hydroxybenzoic acid (4-hydroxy-1-naphthyl-methylene)hydrazide | 342.781 | 3.000 | 4.000 | 3.390 | 95.861 | -2 | -1.267 |
| CP 99-711 | 415.364 | 0.000 | 4.500 | 5.967 | 100.000 | 2 | 0.571 |
| NNC 92-1687 | 300.331 | 3.000 | 5.000 | 1.903 | 77.500 | -2 | -1.547 |
| L-168,049 | 467.792 | 1.000 | 2.250 | 7.393 | 100.000 | 1 | 0.363 |
| NNC 25-0926 | 582.482 | 3.000 | 7.200 | 5.459 | 62.616 | -2 | -1.434 |
| Skyrin | 538.466 | 2.000 | 8.500 | 2.047 | 21.879 | -2 | -3.354 |
| Spirourea | 209.291 | 3.000 | 1.500 | 1.322 | 77.749 | -1 | -0.718 |
| Benzimidazole | 118.138 | 1.000 | 1.500 | 1.320 | 95.861 | 1 | 0.106 |
| STOCK 1N82694 | 483.476 | 2.000 | 6.750 | 3.799 | 80.689 | -2 | -2.219 |
| Recommended ranges: MW (Molecular weight)<500; HBD (Hydrogen Bond Donors): 0 to 5; HBA (Hydrogen Bond Acceptors): 0 to 10; log P (Octanol/water partition coefficient) <5.0; % Oral Absorption >80% High, <25% Poor; CNS Activity -2 Inactive, 2 Active; BBB (Blood Brain Barrier) Partition Coefficient-3.0 to 1.2. | |||||||
Table 3: ADME properties of some already published GCGR antagonists and STOCK1N82694.
Glucagon plays an important role in glucose homeostasis through activation of GCGR. The obstruction of glucagon-GCGR interaction has been notified to control the overproduction of liver glucose. Thus, it can act as a therapeutic target for the treatment of type 2 diabetes mellitus. Various safety, tolerability and immunological concerns related with already documented GCGR antagonists, making them unfit for clinical use motivated us to discover safe compound with acceptable pharmacological properties. We have proposed a human GCGR antagonist based on rational drug design. Docking and Molecular Simulation studies revealed the better binding interaction of STOCK1N82694 with GCGR. STOCK1N82694 is having acceptable pharmacological properties thus it could be a futuristic perspective chemical compound for the treatment of type 2 diabetes mellitus.