Journal of Cell Science & Therapy
Open Access
ISSN: 2157-7013
ISSN: 2157-7013
Research Article - (2017) Volume 8, Issue 5
S-phase-kinase-associated protein 2 (SKP2) plays a crucial role in the tumorigenesis of various human cancers. Imatinib, an inhibitor of BCR-ABL tyrosine kinase, c-kit, and platelet derived growth factor receptors, has been approved for the treatment and investigation of chronic myeloid leukemia, gastrointestinal stromal tumors, and various other solid tumors. However, its efficacy in the treatment of skin squamous cell carcinoma (SSCC) has not yet been investigated. The aim of this study was to investigate whether imatinib has an antitumor effect in human SSCC cells and evaluate its functional relationship with SKP2. To research the effect of imatinib on the growth of A431 SSCC cells, we conducted a proliferation assay and flow cytometry. A western blot assay was performed to verify the function of imatinib in A431 cells and evaluate the underlying molecular mechanism. Imatinib significantly inhibited cell growth and induced cell cycle arrest at the G0/G1 phase. Furthermore, the depletion of SKP2 triggered cell cycle arrest and inhibited colony formation. Mechanistically, imatinib markedly downregulated SKP2 protein expression and subsequently upregulated P21 expression via increased protein stability. In conclusion, imatinib demonstrated an antitumor activity against SSCC cells via the SKP2-P21 signaling axis and targeting SKP2 using imatinib could be a novel therapeutic approach for SSCC that warrants further study.
Keywords: SKP2, P21, Imatinib, Skin squamous cell carcinoma
Many anticancer drugs target genes related to cellular processes. SKP2 is a component of the SCFSKP2 E3 ubiquitin ligase complex; it promotes the ubiquitination and proteasomal degradation of its substrates. Known SKP2 substrates include P27KIP1, P21CIP1, p57KIP2, and E2F1 [1-4], which contribute to the regulation of cellular growth and apoptosis. Several papers have reported that the expression of SKP2 was increased in various cancer tissues and cells as compared with non-cancer tissues and cells. Besides, a strong SKP2 expression correlates with metastasis. Thus, SKP2 is regarded as a prognostic marker for colorectal cancer [5-9]. These observations suggest that as a proto-oncogene, SKP2 plays a critical role in cancer cell processes and is a promising therapeutic target in various human cancers. Recent studies have indicated that the BCR-ABL oncoprotein upregulates the expression of SKP2 in chronic myeloid leukemia (CML) cells and imatinib inhibits BCR-ABL tyrosine kinase activity via the downregulation of SKP2 [10,11].
Skin cancer occurs because of the abnormal growth of skin cells. One type of skin cancer, skin squamous cell carcinoma (SSCC), represents approximately 20% of non-melanoma cases. In 2013, 180,000 ~ 400,000 cases of SSCC were reported in the United States [12]. SSCC usually develops in males aged over 60, and individuals with lighter-colored skin have a higher risk of pathogenesis. SSCC is generally treated by surgical resection and non- surgical treatments such as chemotherapy or radiotherapy. SKP2 protein has been reported to be overexpressed in human oral squamous cell carcinoma samples, and the overexpression of SKP2 has been demonstrated to promote radiation resistance in esophageal squamous cell carcinoma [13,14].
Tyrosine kinase inhibitors bind the hydrophobic pocket of the ATPbinding site of their target tyrosine kinases, blocking their kinase activity. Several tyrosine kinase inhibitors with antitumor effects have been subjected to clinical trials in cancer patients. Imatinib (Gleevec®, STI571) was developed for patients with early-stage CML. Although imatinib was developed to target a specific kinase, namely BCR-ABL, which is responsible for CML, many studies have established that imatinib also inhibits platelet-derived growth factor (PDGF) receptors and c-kit in various human cancers. Matei et al. reported that imatinib inhibits the growth of ovarian cancer cells through inactivating PDGF receptors and AKT [15]. Moreover, imatinib is used in the treatment of various cancers, including gastrointestinal stromal tumors, acute lymphoblastic leukemia, and plexiform neurofibroma [13,16]. The use of imatinib for the treatment of lung cancer, malignant brain tumors, and diverse solid tumors is currently under clinical investigation. However, its efficacy for treating SSCC has yet to be investigated.
In this study, we identified candidate SKP2 inhibitors via a virtual screening method based on the crystal structure of SKP2 and we explored the effect of imatinib, which was one of the candidates identified during the screening process, on the SSCC cell line A431. We found that imatinib inhibited cell growth and induced cell cycle arrest in these SSCC cells through downregulation of SKP2 and the upregulation of P21CIP1. Our data suggested that imatinib, which is an antitumor drug that shows potential for the treatment of SSCC.
Cell culture and reagents
The human SSCC cell line A431 was cultured in Dulbecco’s modified Eagle’s medium. Human lung cancer cell lines (HCC827 and H1650) and the gefitinib-resistant lung cancer cell line HCC827 GR were cultured in Roswell Park Memorial Institute 1640 medium. The human colon cancer cell line HCT116 was cultured in McCoy’s 5A medium. All medium contains 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin and cells were incubated at 37°C with 5% CO2 and 95% humidity. Imatinib, [6]-shogaol, GNF-2, GNF-5, and silybin were purchased from Cayman Chemical (Ann Arbor, MI, USA) and dissolved in dimethylsulfoxide at 20 mM. Gossypetin was purchased from Indofine (Somerville, NJ, USA) and dissolved in dimethylsulfoxide at 50 or 100 mM.
Cell viability assay
Cell viability was measured by the Cell Counting Kit-8 (CCK-8) assay (Dojindo Molecular Technologies, Rockville, MD, USA). In brief, cells were seeded in 96-well plates (2 × 10³ cells/well) and incubated for 18 h with imatinib at 0, 10, 20, or 40 μM. The cells were incubated for 0, 24, 48, 72, or 96 h at 37°C and then incubated for 2 h with CCK-8 solution (10 μL/well). Absorbance was determined at 450 nM using a microplate spectrophotometer (BMG LABTECH, Ortenberg, Germany).
Assay for detection of cell cycle progression
Cells were seeded in 6-well plates (3 × 103 cells/well) for 18 h and treated with the indicated concentrations of imatinib. After 24 h, the cells were harvested and washed with cold phosphate-buffered saline (PBS), then fixed with cold 70% ethanol. The fixed cells were washed with cold PBS and incubated with 10 mg/mL RNase A, 50 μg/mL propidium iodide, and 0.5% Triton™ X-100 in PBS at 37°C for 30 min. Cell cycle stage was analyzed using a FACSC alibur™ flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Immunoblot assay
Cells were lysed with radioimmunoprecipitation assay buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton™ X-100, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) and 1% protease inhibitor (GenDEPOT, Barker, TX, USA). Protein concentrations were measured by DC™ assay (Bio-Rad Laboratories, Hercules, CA, USA). Proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis, electrotransferred to polyvinylidene fluoride membranes, and then incubated with the suitable primary antibody overnight at 4°C. These membranes were then incubated with an appropriate secondary antibody for 1 h at room temperature (20– 25°C) after washing them with Tris-buffered saline containing Tween® 20. The expression of specific proteins was detected using a Davinch-Chemi™ imaging system (Davinch-K, Seoul, Korea). Primary antibodies for SKP2 and P27 and all secondary antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Antibodies against P21, tubulin, CDK-2, cyclin E1, CDK-4, and cyclin D1 were purchased from Cell Signaling Technology (Danvers, MA, USA). Cycloheximide (CHX) and MG132 were purchased from Sigma-Aldrich (St. Louis, MO, USA).
CHX chase analysis
Cells were seeded in 6-well plates and treated with or without imatinib. After 20 h, the cells were treated with CHX (20 μg/mL), which is a eukaryotic protein synthesis inhibitor. At 0, 1, 2, or 4 h following CHX treatment, cells were harvested and their protein expression levels were analyzed. A proteasome inhibitor, MG132 (10 μM) was added 2 h before harvesting the cells for analysis.
Lentiviral particle production and infection
HEK-293T cells were transfected for 24 h with the pLKO.1-sh Mock or pLKO.1-sh SKP2 plasmid and a lentiviral envelope packaging plasmid (pRSV-Rev, pMDLg/pRRE) using the jetPEI™ transfection reagent (Cosmogenetech, Seoul, Korea). Viral particles were collected 24 h after the initial transfection. A431 cells were incubated with viral particles for 48 h in a medium containing polybrene (Sigma-Aldrich). Subsequently, cells were selected by treatment with puromycin (2 μg/mL) for 48 h and the efficacy of SKP2 depletion was validated by immunoblotting.
Soft agar assay for colony formation
Cells (8 × 103) were plated in triplicate on top of a bottom layer with 0.5% agar and 10% FBS and the indicated concentration of imatinib. Cells were suspended in Dulbecco’s modified Eagle’s medium containing 0.3% agar, 10% FBS, and the indicated concentration of imatinib. After four weeks, colonies were counted under a light microscope.
Statistical analysis
All experimental results are presented as the mean ± standard deviation. Significant differences were determined using Student’s ttest. A value of p< 0.05 was considered to represent statistical significance.
Imatinib downregulates SKP2 protein expression and inhibits cell growth in A431 cells
The structure of SKP2 protein was analyzed using a super computer in the Research Collaboratory for Structural Bioinformatics Protein Data Bank. We performed virtual screening using the crystal structure of SKP2 and the chemical structures of compounds in the US Food and Drug Administration (FDA) Drug Databases and Natural Product Databases as inputs [17]. As a result, we identified [6]-shogaol, imatinib, GNF-2, GNF-5, silybin and gossypetin as candidate inhibitors of SKP2. We evaluated the endogenous abundance of SKP2 protein in human lung cancer cell lines (HCC827 and H1650), a gefitinib-resistant lung cancer cell line (HCC827 GR), a human colon cancer cell line (HCT116), and a human SSCC cell line (A431). As shown in Figure 1A, the expression of the SKP2 was higher in A431 cells than in the other cell lines. To explore the SKP2-inhibitory effect of the candidate compounds in A431 cells, the cells were incubated with each candidate compound for 24 h. Our data showed that among the candidates, imatinib, GNF-2, and GNF-5 strongly reduced the abundance of SKP2 protein (Figure 1B). Many studies have demonstrated that imatinib inhibits cell growth in various cancers, but its effect on the growth of SSCC cells has yet to be investigated [18,19]. To explore whether imatinib influences cell growth in SSCC cells, we measured the viability of A431 cells treated with graded concentrations of imatinib for 0, 24, 48, 72, or 96 h. Imatinib significantly inhibited cell proliferation in a time- and dose- dependent manner (Figure 1C). Furthermore, A431 cells treated with imatinib displayed a dosedependent decrease in the abundance of SKP2 protein (Figure 1D).
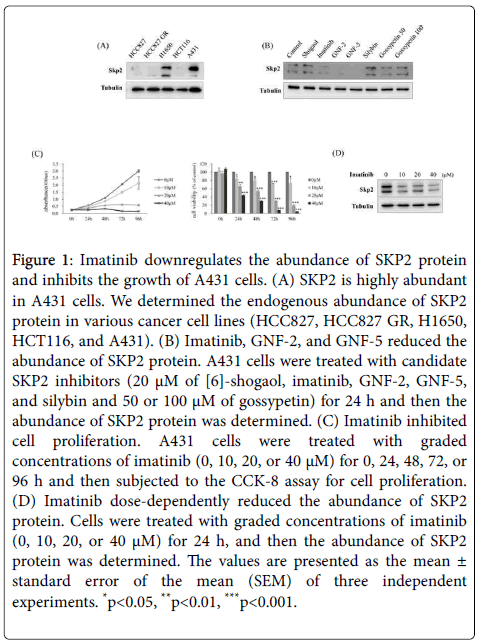
Figure 1: Imatinib downregulates the abundance of SKP2 protein and inhibits the growth of A431 cells. (A) SKP2 is highly abundant in A431 cells. We determined the endogenous abundance of SKP2 protein in various cancer cell lines (HCC827, HCC827 GR, H1650, HCT116, and A431). (B) Imatinib, GNF-2, and GNF-5 reduced the abundance of SKP2 protein. A431 cells were treated with candidate SKP2 inhibitors (20 μM of [6]-shogaol, imatinib, GNF-2, GNF-5, and silybin and 50 or 100 μM of gossypetin) for 24 h and then the abundance of SKP2 protein was determined. (C) Imatinib inhibited cell proliferation. A431 cells were treated with graded concentrations of imatinib (0, 10, 20, or 40 μM) for 0, 24, 48, 72, or 96 h and then subjected to the CCK-8 assay for cell proliferation. (D) Imatinib dose-dependently reduced the abundance of SKP2 protein. Cells were treated with graded concentrations of imatinib (0, 10, 20, or 40 μM) for 24 h, and then the abundance of SKP2 protein was determined. The values are presented as the mean ± standard error of the mean (SEM) of three independent experiments. *p< 0.05, **p< 0.01, ***p< 0.001.
Imatinib induces cell cycle arrest at the G0/G1 phase in A431 cells
SKP2 is a crucial regulator of the cell cycle process that induces the ubiquitination of cyclin- dependent kinase inhibitors including P27KIP1, P21CIP1, and p57KIP2 [20]. Previous studies have reported that imatinib increases the expression of P21CIP1 and P27KIP1 and promotes cell cycle arrest [21,22]. To investigate the mechanism of imatinib-induced cell growth inhibition in SSCC cells, we conducted a cell cycle assay. A431 cells were incubated with imatinib for 24 h and then analyzed by flow cytometry. The accumulation of cells in the G0/G1 phase was observed following treatment with imatinib in a dose-dependent manner. Conversely, imatinib treatment caused a reduction in the proportions of the cell population in the S and G2/M phases (Figure 2A). We measured the abundance of the P21CIP1 and P27KIP1 proteins, which are SKP2 substrates, in A431 cells treated with imatinib for 48 h. As expected, treatment with imatinib upregulated the abundance of P21CIP1 protein in a dose-dependent manner, but that of P27KIP1 protein remained unchanged. The cell cycle-related proteins CDK2 and cyclin E1 were downregulated in A431 cells treated with imatinib (Figure 2B). Our data demonstrated that imatinib inhibits cell growth in SSCC cells through the induction of cell cycle arrest at the G0/G1 phase.
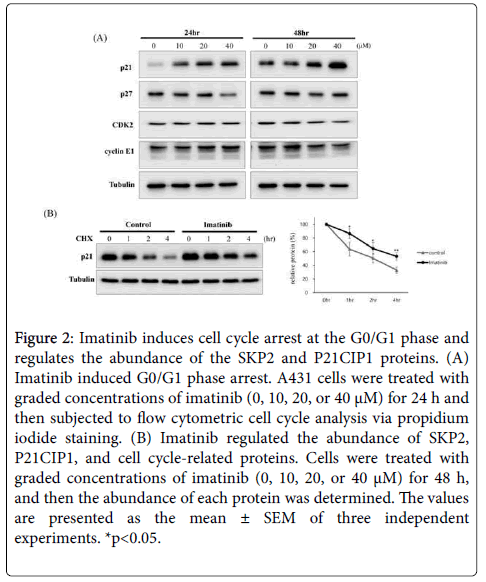
Figure 2: Imatinib induces cell cycle arrest at the G0/G1 phase and regulates the abundance of the SKP2 and P21CIP1 proteins. (A) Imatinib induced G0/G1 phase arrest. A431 cells were treated with graded concentrations of imatinib (0, 10, 20, or 40 μM) for 24 h and then subjected to flow cytometric cell cycle analysis via propidium iodide staining. (B) Imatinib regulated the abundance of SKP2, P21CIP1, and cell cycle-related proteins. Cells were treated with graded concentrations of imatinib (0, 10, 20, or 40 μM) for 48 h, and then the abundance of each protein was determined. The values are presented as the mean ± SEM of three independent experiments. *p< 0.05.
Imatinib upregulates P21CIP1 protein stability via the downregulation of SKP2 protein expression
To further validate the mechanism of the imatinib-induced downregulation of SKP2, A431 cells were treated with imatinib for 24 h in the absence or presence of the cell-permeable reversible proteasome inhibitor MG132 and the protein synthesis inhibitor CHX, and the abundance of SKP2 protein was measured. The abundance of SKP2 protein was reduced in the presence of imatinib, but it was significantly restored by MG132 treatment (Figure 3A). Our results showed that imatinib downregulates the abundance of SKP2 protein by promoting its proteasomal degradation. Additionally, we analyzed the kinetics of P21CIP1 degradation using a CHX chase assay. The stability of P21CIP1 was significantly prolonged in A431 cells treated with imatinib (Figure 3B). To determine whether the change in the abundance of SKP2 protein was responsible for the imatinib-induced increase in P21CIP1 stability, we generated a stable cell line with a depleted abundance of SKP2 protein. For the generation of this cell lines, A431 cells were transfected with five types of short hairpin (sh) RNA targeting SKP2 and the abundance of SKP2 protein was determined in each transfected cell line. Based on the results, we selected shSKP2-1 and shSKP2-3 for the generation of the SKP2- deficient cell line. The generated cell line was treated with imatinib for 24 h, and the abundance of SKP2 and P21CIP1 proteins was measured. According to our results, imatinib treatment upregulated the abundance of P21CIP1 protein more strongly in the SKP2-deficient cell line than in non-SKP2- deficient cells (Figure 3C). Taken together, our data showed that imatinib treatment upregulated the stability of P21CIP1 protein by promoting SKP2 degradation.
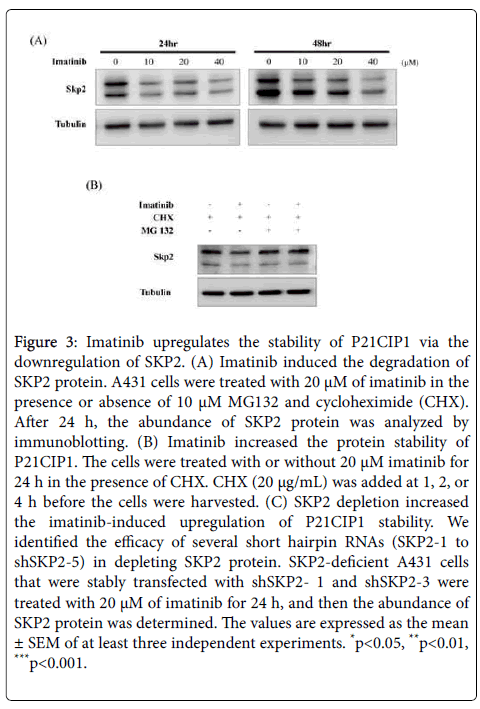
Figure 3: Imatinib upregulates the stability of P21CIP1 via the downregulation of SKP2. (A) Imatinib induced the degradation of SKP2 protein. A431 cells were treated with 20 μM of imatinib in the presence or absence of 10 μM MG132 and cycloheximide (CHX). After 24 h, the abundance of SKP2 protein was analyzed by immunoblotting. (B) Imatinib increased the protein stability of P21CIP1. The cells were treated with or without 20 μM imatinib for 24 h in the presence of CHX. CHX (20 μg/mL) was added at 1, 2, or 4 h before the cells were harvested. (C) SKP2 depletion increased the imatinib-induced upregulation of P21CIP1 stability. We identified the efficacy of several short hairpin RNAs (SKP2-1 to shSKP2-5) in depleting SKP2 protein. SKP2-deficient A431 cells that were stably transfected with shSKP2- 1 and shSKP2-3 were treated with 20 μM of imatinib for 24 h, and then the abundance of SKP2 protein was determined. The values are expressed as the mean ± SEM of at least three independent experiments. *p< 0.05, **p< 0.01, ***p< 0.001.
The depletion of SKP2 protein enhances antitumor effects by increasing imatinib sensitivity
To determine whether the depletion of SKP2 protein affected imatinib-induced cell cycle arrest we performed a cell cycle assay. An increase in the proportion of the cell population in G0/G1 phase was observed following SKP2 depletion by sh-SKP2 and treatment with imatinib (Figure 4A). Furthermore, the depletion of SKP2 dramatically decreased the abundance of CDK2 and cyclin E1 following incubation with imatinib (Figure 4B). Next, we conducted a colony formation assay to detect the malignant transformation of the SSCC cells. The data showed that the depletion of SKP2 significantly attenuated the colony formation ability of the SSCC cells. Notably, the depletion of SKP2 in combination with imatinib treatment inhibited the colony formation ability more strongly than imatinib alone or SKP2 depletion alone did (Figure 4C). These results suggest that SKP2 depletion increased the sensitivity of A431 SSCC cells to the imatinib-induced inhibition of cell growth.

Figure 4: The depletion of SKP2 promotes imatinib-induced cell cycle arrest and the inhibition of colony formation. (A) SKP2 depletion increases imatinib-induced G0/G1 phase arrest. SKP2- depleted cells were treated with or without imatinib (20 μM) for 24 h and then subjected to flow cytometric cell cycle analysis via propidium iodide staining. (B) SKP2 depletion increased the imatinib- induced upregulation of P21CIP1 and reduced the abundance of its target proteins. SKP2- depleted A431 cells were treated with or without imatinib (20 μM) for 24 h. A western blot analysis was conducted to determine the abundance of the target proteins. (C) SKP2 depletion increased the imatinib-induced inhibition of colony formation. Representative images are shown that reveal the clonogenic ability of SKP2-depleted cells treated with or without imatinib (20 μM) for 4 weeks. The values are expressed as the mean ± SEM of at least three independent experiments. *p< 0.05, **p< 0.01, ***p< 0.001.
We determined that imatinib inhibited cell growth and induced cell cycle arrest in A431 SSCC cells. Especially, our results demonstrated the existence of a relationship between the SKP2-P21CIP1 axis and the antitumor effect of imatinib. The downregulation of SKP2 and subsequent stabilization of P21CIP1 mediated the inhibition of cell growth and delay of cell cycle progression that were induced by imatinib treatment. Moreover, the depletion of SKP2 in combination with imatinib treatment triggered cell cycle arrest and the long-term suppression of cell proliferation.
Imatinib is a tyrosine kinase inhibitor for BCR-ABL, PDGF receptors, and c-kit [23,24]. It is also used for the treatment of gastrointestinal stromal tumors that overexpress c-kit as well as CML [25]. A431 cells are known to express epidermal growth factor receptors, but they do not highly express the previously characterized targets of imatinib such as BCR-ABL, c-kit, and PDGF receptors. A recent study showed that imatinib accelerates the activation of epidermal growth factor tyrosine kinase receptors despite causing a decrease in cell number [26]. Previous studies have demonstrated that imatinib significantly inhibits the growth of head and neck squamous cell carcinoma cell lines [27]. Moreover, imatinib induced an increased radiosensitivity of human squamous cell carcinoma and glioblastoma cells [28]. Baskaynak et al. reported that SSCCs were observed in two patients who received imatinib therapy for CML [29]. In contrast, Kawakami et al. reported the remission of an SSCC by imatinib treatment in a patient with CML [30].
The degradation of Skp2 by APC/Ccdh1 has already been demonstrated, and several studies have demonstrated the increased expression of Cdh1 by Imatinib [31] and the signaling axis of Imatinib-induced Cdh1-Skp2-p27 [32]. In this study, we also examined whether Imatinib-induced downregulation of Skp2 is associated with the expression of Cdh1. As a result, the increase of Cdh1 expression by imatinib was not confirmed, rather it was confirmed as reduced by imatinib. I did not check the effect of imatinib on another APC/C activator, Cdc20, but it is not related to the downregulation of Skp2 by imatinib as confirmed in this study, because it has a complementary role with Cdh1.
SKP2 is highly expressed in various human cancers and causes the degradation of the tumor suppressor proteins P21CIP1 and P27KIP1 [14,33]. It has been indicated that SKP2 downregulation has an antitumor effect against various cancer cell lines [8,34]. Agarwal et al. have shown that SKP2 is involved in leukemogenesis by BCR-ABL and promotes oncogenesis in vivo [35]. Liu et al. demonstrated that imatinib induces the downregulation of SKP2 and the upregulation of nuclear CDH1 in gastrointestinal stromal tumor cells [32].
Several studies have demonstrated that imatinib upregulates P21CIP1, leading to the inhibition of cell proliferation. For example, an increase in the abundance of P21CIP1 protein and G0/G1 arrest following imatinib treatment was reported for multiple myeloma cells [22]. In addition, imatinib inhibits cell proliferation and induces cell cycle arrest at the G0/G1 phase through the inactivation of extracellular signal-regulated kinase, the blocking of AKT signaling pathways, and the upregulation of P21CIP1 in human breast stromal fibroblasts [21]. In the present study, we demonstrated that imatinib significantly reduced the abundance of SKP2 protein by promoting its degradation while stabilizing P21CIP1. Additionally, the depletion of SKP2 enhanced cell cycle arrest and significantly inhibited the anchorage-independent cell growth induced by imatinib.
In conclusion, our study found that imatinib exerts its antitumor activities, including the suppression of cell proliferation and the induction of cell cycle arrest, partly via the inhibition of the SKP2- P21CIP1 axis, and these results suggested that targeting SKP2 with imatinib could be a novel therapeutic approach for SSCC that warrants further study.
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (No. 2014R1A6A3A04059193); by an NRF grant funded by the Korean government (MSIP) (No. 2015R1A5A6001906); by a grant of the Korea Health technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (No. HI16C1501); and by the Basic Science Research Program through the NRF funded by the Ministry of Education, Science and Technology (No. 2014R1A1A2059813).