Journal of Plant Biochemistry & Physiology
Open Access
ISSN: 2329-9029
ISSN: 2329-9029
Research Article - (2018) Volume 6, Issue 2
Indole-3-acetic acid (IAA) is the primary form of auxin in plants and several IAA biosynthetic pathways have been previously proposed but remain genetically uncharacterized. One of the existing pathways is the indole-3-pyruvic acid (IPyA) pathway, which is known to regulate key developmental processes such as apical hook formation and shade avoidance. Recent studies suggest the existence of the pathway in higher plants but are unverified due to the elusive nature of IPyA in vitro. Extending on these recent advances, this research was aimed at investigating aspects of IPyA-dependent auxin biology in Pisum sativum (pea) using reverse genetics. Consequently, using a reverse genetic approach, called TILLING, the PsTAR2 gene was mutated in order to study firsthand the downstream effects of IPyA deficiency.
The procedure resulted in isolating two novel PsTAR2 (IPyA) mutant lines consisting of a missense mutation (PsTAR2 4280) and a highly desired knockout mutation (PsTAR2 918).
The novel mutants are anticipated to be indispensable to future IPyA-auxin investigations in higher plants.
Keywords: Auxin; Indole-3-acetic acid; Indole-3-pyruvic acid; Genes
AA: Acetic Acid; ACN: Acetonitrile; DNA: Deoxyribonucleic Acid; cDNA: Complementary DNA; EMS: Ethylmethane Sulfonate.
Indole-3-pyruvic acid (IPyA) has long been hypothesised as an intermediate in auxin biosynthesis. However, only recently are its functions in auxin biology being slowly unravelled. Tao and Ferrer conducted a genetic screen for Arabidopsis mutants that were defective in shade avoidance response, a process in which plants elongate in response to altered light conditions, and isolated a perturbed gene (TAA1) that encoded a PLP (pyridoxal-5-phosphate)-dependent Trpaminotransferase enzyme [1]. The TAA1 mutants were found to contain lesser global amounts of free IAA [2] and showed decreased IAA biosynthesis rates in response to shade conditions [1]. Transcriptional analysis of TAA1 mutants revealed several auxin-inducible genes that were down-regulated. Two other independent genetic screens involving ethylene-insensitivity and temperature-dependent hypocotyl elongation, again led to the identification of TAA1 as WEI8 (WEAK ETHYLENE INSENSITIVE 8) and its close homologs TAR1, TAR2, TAR3, TAR4 [1-4]. The wei8 mutants shared similar impeded auxin biosynthetic activity as TAA1 mutants, leading to reduced IAA levels. The condition was found to be partially rescued by introducing exogenous IAA, confirming impeded auxin biosynthetic activity [1]. Simultaneous inactivation of TAA1 and its two close homologs TAR1 and TAR2 led to defects that closely resembled auxin deficiency, similar to the well-known auxin mutants monopteros and pinoid [5]. The collective findings led to the linking of taa1 /tar2 to IPyAdependent auxin biosynthesis, and IPyA-dependent auxin to shade responses. The Trp-aminotransferase (TAA1/TAR2 ) enzyme is widespread throughout the plant kingdom [4], and the IPyA pathway is highly conserved, suggesting high significance in auxin biosynthesis. Yet the pathway remains uncharacterised in higher plants due to the elusive and unstable nature of IPyA in vitro, and the functional redundancy that exists in the pathway.
Further elucidation of IPyA-dependent auxin biosynthesis in higher plants involves characterising the molecular components that constitute the pathways, in particular the genes that encode the enzymes. This research addresses this issue by inducing polymorphisms in the PsTAR2 gene, the AtTAR2 homolog in peas, using a reverse genetic approach called Targeted Induced Local Lesions in Genomes (TILLING). This approach would enable studying the direct down-stream effects of IPyA deficiency, and create novel IPyA auxin biosynthetic mutants in the process. Explained below is a brief description of the reverse genetic procedure that was used to attain the desired mutants.
TILLING is a chemical mutagenesis procedure that utilises ethylmethane sulfonate (EMS) to induce inheritable single nucleotide polymorphisms (SNPs) at a moderate to high density throughout the host genome. EMS is an organic compound that induces point mutations by nucleotide substitution, primarily by alkylation on the O6 (oxygen at position 6) position of guanine (G) leading to GC→AT transitions [6]. Apart from the relative mutational uniformity, the process commonly yields an allelic series of mutations in nontransformable species, such as Pisum sativum (peas). The mutations are detected as heteroduplexes, after endonuclease digestion, which form between wild-type (WT) and mutant DNA strands. A database (UTILLdb) of the mutagenized pea cultivar Cameor is publicly available in the web interface with all the available mutations. On request of specific mutant lines, the polymorphisms are confirmed through sequencing and the lines are dispatched accordingly. Coupled with the recent advent of high-throughput screening for SNPs, TILLING is a powerful and efficient mutagenesis procedure for species that are innately recalcitrant towards traditional agro bacterial transformation (T-DNA), and where an allelic mutant series is essential to characterise complex gene functions and interactions. This procedure also serves as a novel exercise in TILLING the pea genome for hormone-related genes, and assesses the efficiency of the procedure towards isolating the perturbed gene of interest.
Mutagenesis
Ethylmethane sulfonate (EMS) was diluted in deionised water to a mutational dosage of 16-24 mM as described [7]. Cameor (pea cultivar) seeds were soaked in the EMS solution and grown in glasshouse conditions. The temperature was maintained at 14°C at night and 30°C during the day, with a photoperiod of 16 hours. The M1 plants were harvested for seeds (M2), and the M2 generation was propagated in similar glasshouse conditions as described [7].
DNA extraction and pooling
Four pea leaf discs (10 mm) were collected from the M2 plants in 96-well plates containing two steel beads (4 mm) per well, and the tissues were ground using a bead mill. DNA was isolated using the DNeasy 96 Plant Kit (Qiagen, Germany). The genomic DNA was quantified on a 0.8% agarose gel using λ DNA (Invitrogen, USA). DNA samples were diluted tenfold and normalised to ensure all lines were equally represented in pools (eightfold in 96-well format).
Mutation detection
The DNA pools were PCR amplified based on nested-PCR and universal primers as described [7]. The first PCR amplification was a standard PCR reaction using target-specific primers with 4 ng of pea genomic DNA. 1 μl of the first PCR served as a template for the second nested PCR amplification, using gene-specific inner primers (TAA1) carrying a universal M13 tail (CACGACGTTGTAAAACGAC for forward primers; GGATAACATTTCACACAGG for reverse primers), in combination with M13 universal primers, M13F700 (CACGACGTTGTAAAACGAC) and M13R800 (GGATAACAATTTCACACAGG), labelled at the 5’ end with infrared dyes IRD700 and IRD800 (LI-COR®, Lincoln, NE, USA), respectively. The PCR was carried out using 0.1 μM of each primer, using the following a two-step cycling program (94°C for 2 min, 10 cycles at 94°C for 15 sec, 72°C for 1 min, 5 min at 72°C). The procedure leads to the formation of heteroduplexes between the wildtype (WT) and mutated DNA strands due to mismatches. The mismatches were digested using the mismatch specific endonuclease (ENDO1) in buffers as described [8]. The digested PCR products were ran on a polyacrylamide gel, and any cleavage by the enzyme denotes a potential mutation in the pool. The pool was then screened for the polymorphism and the mutant was identified. The use of two channelled dyes that labels 5’ and 3’ end differently enables the mapping of the mutation to approximately 1 kb, which was then confirmed by sequencing.
Mutant acquisition
The known PsTAR2 full-length sequence was used to search the UTILLdb (URGV TILLING database) database for available Cameor mutants in the PsTAR2 gene. Mutant lines were chosen based on SNPs in conserved regions (sequence alignment with Medicago truncatula and Arabidopsis ) of the gene (Figure 1 and Table 1).
| Muttion | Bse Position1 | Protein Position | Type of Muttion | Fmily | No of seeds (M3)2 | Level of genetic conservtion |
|---|---|---|---|---|---|---|
| 1 | C501T | P20S | Missense3 | 4132 | 4 | Low |
| 2 | G542 | W33* | Nonsense* | 918 | 4 | High |
| 3 | G672 | 77T | Missense3 | 3721 | 4 | High |
| 4 | C696T | P85S | Missense3 | 4280 | 4 | High |
| 5 | G720 | D93N | Missense3 | 3087 | 6 | High |
| 6 | G762 | G107R | Missense3 | 3930 | 4 | High |
Table 1: Position and type of mutations of the acquired lines. Colour corresponds to the mutations in the cameor genomic sequence shown above. Where, 1 from beginning of the partial genomic sequence, 2 seeds derived from M2 (Mutagenised generation 2) parents, thus M3, 3 substitution of a different amino acid, *encodes a stop codon.

Figure 1: Partial amplified genomic Cameor sequence. Mutations are highlighted in colour and correspond to the mutant lines in the table below. Highlighted areas represent introns, while the conserved exons are plain. Underlined bases represent the genespecific primers that were used to amplify the genomic DNA.
Plant growth conditions
Seeds from the six mutant lines were cautiously nicked in the testa, while avoiding the emerging radicle. They were treated with Thiram® fungicide for approximately a minute. Standard pots were semi-filled with vermiculate and gravel (1:1) and filled with sterilised soil. The treated seeds were sown in the pots at a depth of 2 cm. The pots were well watered and placed in quarantine (PC2) glasshouse conditions. An average temperature of 18°C at night and 22°C during the day was maintained with a 16 h photoperiod.
An emerging leaflet (~100 μg) was flash-frozen (liquid nitrogen) and shaken with a carbide bead in a bead mill. The DNeasy mini Plant Kit (Qiagen, Germany) was used to extract the DNA following the given protocol. DNA levels were measured on a spectrophotometer (NanaDrop™, USA) when deemed necessary. The genomic DNA (5 μl neat unless specified) was amplified using PCR (94°C for 15 sec, 58°C for 15 sec, 1 min at 72°C) with F1 and 249r Oligo primers (Table 2), which were designed in Primer3™ software and obtained from GeneWorks™ (SA, Australia). 1-2%Polyacrylamide gels were used in trial runs to check the primer and PCR efficiency. The PCR products were purified using a Wizard® SV gel and PCR clean-up system (Promega, WI, USA) following the given protocol. The cleaned PCR products were sent along with primers F3 and r3 (Table 2), and sequenced (genotyped) at Macrogen™ (South Korea). The resulting DNA sequence chromatograms were analysed for the mutations in the Sequencher® software (Michigan, USA). Genotyping was usually based on sequencing unless specified.
| Abbreviated name | Oligo Name | Sequence (5' - 3') |
|---|---|---|
| F1 | tillingcamPsTAR2 F1 | CTC ATG CAA GCT CAA CCA ATC AAC G |
| 249r | tillingcam PsTIR5’ 249r PCR/Seq | AGT TTA GCT CTA TGA ACT TGG TC |
| F3 | tillingcamPsTAR2 F3 PCR/Seq | TAT TCG TGT CGT GTC TGG |
| r3 | tillingcamPsTAR2 r3 PCR/Seq | GTT ACG GTT CTA AGG AG |
Table 2: List of primers used in the sequencing procedure. Primers F1 and 249r were used to amplify the PsTAR2 gene, and primers F3 and r3 were sent with the amplified PCR products for sequencing.
Error bars represent standard error of the mean (n ≥ 3). Samples with different letters and the corresponding same post-fixed digits are significantly different based on two-tailed t-tests assuming equal variance (P<0.05). No letters indicate statistical similarity.
Mutagenised Generation 3 (M3)-several phenotypic segregation
During growth of the M3 population several phenotypes were observed with characteristic shortened internodes at the early developmental stages (2-4 weeks). A plethora of morphological characteristics and defects were observed later at various developmental stages of the plant, but no segregation patterns (Mendelian ratios) were apparent in or between mutant families. The plants were genotyped through sequencing, and there appeared to be no correlation between the morphological defects (phenotypes) and the mutations in the PsTAR2 gene (TAR 2) (Figure 2).

Figure 2: Distinct phenotypes observed in the M3 generation. Heterozygous (TAR2/tar2) mutant plants (right of picture) compared with wild-type cameors (left of picture). Where, (A) Highly branching-4280/1, (B) Pale-3721/4, (C) Short-4280/3,(D) Developmentally defective-3087/4.
Characteristics also differed between the same allelic mutants between the families. For example, the heterozygotes shown (Figure 2) differed markedly in their appearance and architecture. Additionally, the homozygous dominant mutants (TAR2/ TAR2 ) on average did not resemble the WT plants in structure (data not shown). The observations led to the mutant phenotypes being attributed to nonspecific mutations, rather than the genetic condition of the PsTAR2 gene. Two homozygous recessive mutants (tar2/tar2 ) were obtained (3087/1 and 3087/2) from this generation, and were developmentally viable (appendix 1) denoting that the PsTAR2 mutation was non-lethal to the plant’s development.
Later in development (10-15 weeks), several mutant lines proved to be sterile, developmentally inviable, or produced inviable seeds. The lethal phenotypes, as mentioned, were not found to be associated with the PsTAR2 mutation. Hence, plants that were inviable or sterile were henceforth not propagated in order to eliminate non-specific lethal mutations in host genomes. Moreover, the selection of M4 was based on the analysis of the type of polymorphism the individual mutant lines carried.
Mutagenized Generation 4 (M4)-mutant family 918 and 4280 isolation
Lines from two mutant families (918 and 4280) were chosen, based on factors that determine the protein activity, and the genotype of the PsTAR2 gene (see appendix 1). These lines were propagated and formed the M4 generation. The only knockout mutation 918 was reanalysed in order to attain a clearer understanding of the phenotype of the truncated PsTAR2 protein, so as to further limit the number of mutant lines that have to be propagated (due to spatial constraints involving PC2 quarantine).
The 918 polymorphism was sequence aligned (amino acid/protein) with several species, such as Arabidopsis and Medicago (accession nos. AT4G24670 and Medtr3g114550) and was found to encode the stop codon prior (5’ end) to a highly conserved catalytic site. The catalytic site consists of a lysine base which preferentially binds to the co-factor PLP enabling catalytic activity. The 918 mutation excludes this site and prevents catalytic activity. The PsTAR2 918 sequence lengths was also found to be considerably reduced by 227 amino acids, in a 275 amino acid long PsTAR2 protein, thus ensuring total inactivity of the protein (Figure 3).

Figure 3: Protein/amino acid sequence of the PsTAR2 gene. Highlighted in blue is a highly conserved catalytic site and prior to this site is the 918 polymorphism, which is inferred to encode a truncated protein with no activity. Highlighted in grey is the conserved pyridoxal-5'-phosphate (PLP) binding site, and the 4280 mutation that is inferred to perturb the activity of this conserved domain. Where, M -Methionine (start codon), *-918 (null mutation), *-Stop codon, K-Catalytic site, A-4280 (missense mutation).
Clearly a drawback of the mutagenesis procedure was the nonspecific mutations that were segregating along with the gene of interest. It was evident in this study that the chemical mutagenesis procedure gives rise to numerous non-specific mutations with strong phenotypes. Theoretically, every backcross reduces homozygosity in the mutant loci by 50% thus two to three backcrosses were expected to alleviate any non-specific mutations in the 918 family to an acceptable level. Therefore, the healthiest 918 heterozygotes (TAR2/tar2 ) were backcrossed to WT (un-mutagenised) Cameors as no homozygous recessive (tar2/tar2 ) 918’s were detected in the M3 generation (see appendix 1).
Family 918-IpyA knockout
A high degree (55%) of developmental inviability was observed in the 918 seeds of the M4 generation (derived from M3). Of the 11 M4 seeds that were planted, 5 displayed severe developmental disorders leading to premature death, and 1 was incapable of germination. On genotyping the lethal phenotype, the 918 mutation was found to roughly correlate at 88%, with dominants being relatively healthy, and some heterozygotes sharing a short and sterile phenotype, and recessives being aborted at seedling stage. However, this theory was contradicted by a homozygous recessive (tar2/tar2 -line 918/1/5) displaying a normal phenotype (see appendix 2 and 3). Moreover, dissimilarities were found between the backcrossed heterozygotes (WT × mutant) and the normal heterozygotes (mutant × mutant). Thus, the lethal phenotype was suggested to be caused by a non-specific mutation or mutations, further stressing the relevance of backcrossing. The backcrossed 918s (M4) now heterozygous in condition (TAR2/ tar2 ), displayed relatively normal phenotypes (data not shown) after a single round of backcrossing. As a result, the backcrossed heterozygotes were favoured as the suitable population for propagation, with the possibility of one more round of backcrossing pending, in order to effectively eliminate most non-specific mutations.
Family 4280-IPyA sub-functional mutant
Unlike the 918 family, the 4280 families possessed a high level of viability (100%) and fertility (70%), denoting a healthier family (fewer non-specific mutations). The 4280 mutation was sequence aligned with Arabidopsis and Medicago (accession nos. AT4G24670 and Medtr3g114550, protein ID NP_567706.1) and was inferred to affect a conserved pyridoxal-5'-phosphate (PLP) binding site (Figure 3). As mentioned before, PLP is a co-factor that determines protein activity. Thus, any modification to its binding site would considerably deter catalytic activity of the protein. Thus, three 4280 lines (12 seeds from 4280/1, 5 seeds from 4280/3, and 29 seeds from 4280/4) were propagated and formed the majority of the M4 generation.
The M4 lines displayed various phenotypic traits that were segregating in a non-Mendelian fashion, but two traits, namely height and colour, were bimodal in their segregation patterns (Figure 4a and b).
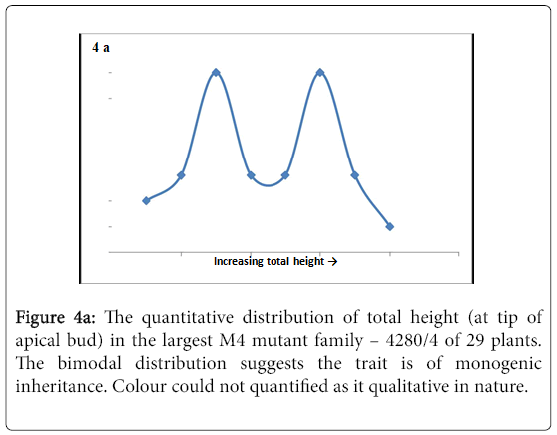
Figure 4a: The quantitative distribution of total height (at tip of apical bud) in the largest M4 mutant family – 4280/4 of 29 plants. The bimodal distribution suggests the trait is of monogenic inheritance. Colour could not quantified as it qualitative in nature.

Figure 4b: The two traits that were segregating independently in the family 4280/4. A-Short phenotype (hh) (right of picture) compared to a WT (left of picture) B- Pale phenotype (cc) (left of picture) compared to WT (right of picture).
On genetic analysis it was revealed that the two traits were monogenic in inheritance (Table 3). Linkage analysis of the two traits and PsTAR2 revealed linkage between colour and PsTAR2 (Table 3). The linkage led to the possibility of colour either being linked to or caused by the PsTAR2 mutation, which remains to be tested. From the literature it was proposed that auxin would be strongly associated with plant height, thus height was further investigated through quantitative measurements on internode lengths and total height in the 4280 families consisting of an allelic series of mutants.
| Trait | Chi-square (X2) | Critical value (P) | Linkage with PsTAR2 |
|---|---|---|---|
| Height | 0.103 | >0.05 | >0.05 |
| Colour | 0.56 | >0.05 | <0.05 |
Table 3: Contingency tests for the traits - height and colour in the 4280/4 (M4) family consisting of 29 plants. The test was based on the assumption of monogenic inheritance, and the observed P-values confirm the hypothesis. Linkage analysis revealed independent assortment between height and PsTAR2 , but not between colour and PsTAR2 .
There is no trend in the scatters further denoting that height and PsTAR2 are not likely to be linked (Figure 5). Given that the 4280 polymorphism is a missense mutation (encodes a leaky protein) there is a still potential for the protein to possess catalytic activity. This may in essence be contributing to a subtle phenotype that is visually indistinguishable. Alternatively, the PsTAR2 mutation may be compensated for by one of the several auxin biosynthetic pathways.
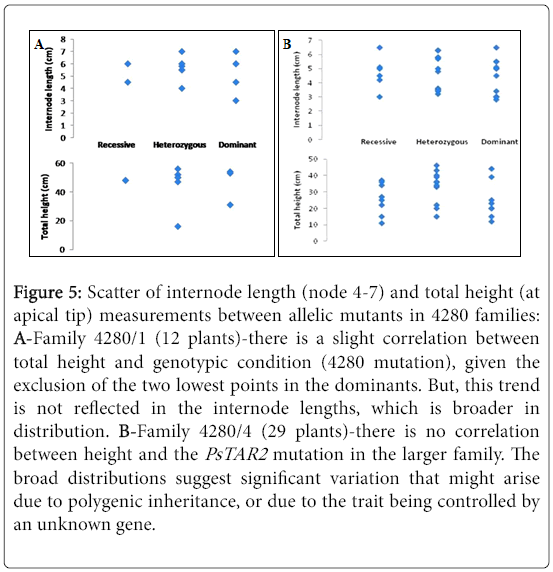
Figure 5: Scatter of internode length (node 4-7) and total height (at apical tip) measurements between allelic mutants in 4280 families: A-Family 4280/1 (12 plants)-there is a slight correlation between total height and genotypic condition (4280 mutation), given the exclusion of the two lowest points in the dominants. But, this trend is not reflected in the internode lengths, which is broader in distribution. B-Family 4280/4 (29 plants)-there is no correlation between height and the PsTAR2 mutation in the larger family. The broad distributions suggest significant variation that might arise due to polygenic inheritance, or due to the trait being controlled by an unknown gene.
Mutagenised Generation 5 (M5)-backcrossing
The backcrossed 918s were displaying relatively high germination rates (90% data), signifying the absence of the non-specific lethal mutations that plagued the initial parental generations (M3 and M4). The careful selection procedure in M3 and M4 has led to the isolation of two mutant lines (918 and 4280) that are inferred to range in functional activity (missense and knockout function). This would be especially being useful in auxin assays, which require a series of mutants that vary in functionality.
The novel auxin biosynthetic mutant strains are allelic in nature, which facilitates studying complex gene functions and interactions. Furthermore, an allelic series of mutants representing independent mutations would reduce the need for lengthy backcrossing programs as related phenotypic variants would strongly associate the phenotype with the gene [6].
918 on a tall background-knockout isolation
From the backcrossed 918 family it was surprising to observe that PsTAR2 918 lacked an effect on plant height since auxin synthesis is known to be strongly associated with height. Further, the presence of ambiguous variations in height in the same mutant lines (918s) raised questions about the parental line (Cameor) used in the study. A possible explanation of the unclear phenotypes was that Cameors were dwarf (le-1) and thus there are restrictions in the height reductions that could be achieved. It was possible that the knockout mutation would be visibly observable on a taller background (cultivar).
Genotyping through sequencing proved time-consuming and expensive, thus an alternative method that could be performed was formulated. This involved closely studying the two polymorphisms (4280 and 918) that were being propagated in the M4 and M5 generations and analysing them for Restriction Fragment Length Polymorphisms (RFLP). RFLPs are differences in restriction fragment lengths caused by SNPs that create or abolish restriction endonuclease recognition sites in PCR amplicons. The 918 and 4280 mutant sequences were analysed in the NEBcutterV2.0® software (Biolabs, England) to find possible restriction enzymes that were capable of selectively differentiating the allelic sequences (Figure 6).

Figure 6: Partial linear sequence of the PsTAR2 gene with all restriction sites and their corresponding restriction enzymes. The enzyme BseRI specifically recognises the 918 mutant sequence relative to the WT: A-Wild-type (WT) linear sequence. B-Mutant (918) linear sequence. Where, Restriction enzyme BseRI recognition site: 5'... G A G G A G (N)10 ... 3', 3'... C T C C T C (N)8 ... 5'
Using the mentioned PCR conditions (see materials and methods) the PsTAR2 gene was amplified, and incubated at 37°C for less than an hour (to prevent non-specific endonuclease activity) in appropriate buffers containing 4 U/μl of BseRI. The digested fragments were run on a 2% polyacrylamide gel (Figure 7).
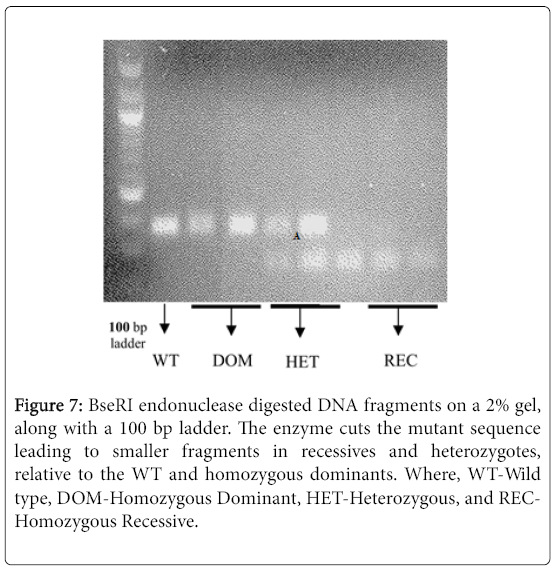
Figure 7: BseRI endonuclease digested DNA fragments on a 2% gel, along with a 100 bp ladder. The enzyme cuts the mutant sequence leading to smaller fragments in recessives and heterozygotes, relative to the WT and homozygous dominants. Where, WT-Wild type, DOM-Homozygous Dominant, HET-Heterozygous, and RECHomozygous Recessive.
The fragments were of expected sizes but differed slightly from the anticipated pattern. A heterozygous condition was expected to lead to three bands, and the recessive condition with two bands, but the smaller fragments were not visible due to limited clarity of the gel (Figure 7). An inferred heterozygote (Figure 8) appeared homozygous recessive in band pattern, but given the sample was a back-crossed L107 (TAR2/tar2 ) this was highly unlikely. The primers were initially designed on Cameor genomes and could be non-specific to the tall lines (L107), in which case there is amplification of the recessive Cameor strand and not the corresponding L107 strand, thus imitating a recessive condition of the sample. Further optimisation, such as increasing PCR efficiency and reducing non-specific endonuclease activity, is necessary to see clear patterns that are characteristic of the allelic conditions. Nonetheless, the bands segregate distinctly with respect to the allelic condition of the 918 mutation, thus proving to be a robust genotypic test for the knockout mutation.

Figure 8: Schematic representation of the RFLP procedure for the 4280 polymorphism using dCAP primers, which were used to introduce two SNPs based on the 4280 mutation. Creating two recognition sites, and subsequent digestion of the PCR products using specific endonucleases, HinfI and AflIII. Predicted bands of the RFLP products based on lengths of digested PCR fragments.
4280 polymorphism ambiguity
On sequence analysis of the 4280 polymorphism, no specific restriction enzymes were found to selectively differentiate the mutant and WT sequences (i.e., no restriction sites were present that differed between the two alleles). A modification of RFLP was created to acquire the desired polymorphic specificity, in which the PCR primers were redesigned (dCAPS FINDER 2.0®) to forcibly induce new polymorphisms based on the target 4280 mutation and thus creating recognition sites in the alleles (Figure 8).
A pilot study was conducted with amplification (see materials and methods) and incubation at 37°C for less than 24 hours in appropriate buffers containing 4 U/μl of HinfI. The digested fragments were run on a 2% polyacrylamide gel. However, unlike the 918 mutation, the 4280 polymorphism RFLP analysis proved relatively unyielding (data not shown) possibly due to a combination of factors, including primer inefficiency, non-specific endonuclease activity, mismatch intolerance, or background mutations in the 3’ end primer region. A dominant and a recessive sample showed clear band patterns (data not shown) suggesting the amplification and the digestion were successful. However, the same could not be said for the other samples. The primer inefficiency and mismatch intolerance could be greatly reduced through reducing annealing temperatures, since base pair specificity is a direct function of temperature. As for non-specific enzymatic activity, the period of incubation can be reduced given the above modifications prove ineffective.
Genotyping through sequencing proved time-consuming and expensive, thus an alternative method that could be performed was formulated. This involved closely studying the two polymorphisms (4280 and 918) that were being propagated in the M4 and M5 generations and analysing them for Restriction Fragment Length Polymorphisms (RFLP). RFLPs are differences in restriction fragment lengths caused by SNPs that create or abolish restriction endonuclease recognition sites in PCR amplicons. The 918 and 4280 mutant sequences were analysed in the NEBcutterV2.0® software (Biolabs, England) to find possible restriction enzymes that were capable of selectively differentiating the allelic sequences (Figure 6).
Figure 6: Partial linear sequence of the PsTAR2 gene with all restriction sites and their corresponding restriction enzymes. The enzyme BseRI specifically recognises the 918 mutant sequence relative to the WT: A-Wild-type (WT) linear sequence. B-Mutant (918) linear sequence. Where, Restriction enzyme BseRI recognition site: 5'... G A G G A G (N)10 ... 3', 3'... C T C C T C (N)8 ... 5'
Using the mentioned PCR conditions (see materials and methods) the PsTAR2 gene was amplified, and incubated at 37°C for less than an hour (to prevent non-specific endonuclease activity) in appropriate buffers containing 4 U/μl of BseRI. The digested fragments were run on a 2% polyacrylamide gel (Figure 7).
Figure 7: BseRI endonuclease digested DNA fragments on a 2% gel, along with a 100 bp ladder. The enzyme cuts the mutant sequence leading to smaller fragments in recessives and heterozygotes, relative to the WT and homozygous dominants. Where, WT-Wild type, DOM-Homozygous Dominant, HET-Heterozygous, and RECHomozygous Recessive.
The fragments were of expected sizes but differed slightly from the anticipated pattern. A heterozygous condition was expected to lead to three bands, and the recessive condition with two bands, but the smaller fragments were not visible due to limited clarity of the gel (Figure 7). An inferred heterozygote (Figure 8) appeared homozygous recessive in band pattern, but given the sample was a back-crossed L107 (TAR2/tar2 ) this was highly unlikely. The primers were initially designed on Cameor genomes and could be non-specific to the tall lines (L107), in which case there is amplification of the recessive Cameor strand and not the corresponding L107 strand, thus imitating a recessive condition of the sample. Further optimisation, such as increasing PCR efficiency and reducing non-specific endonuclease activity, is necessary to see clear patterns that are characteristic of the allelic conditions. Nonetheless, the bands segregate distinctly with respect to the allelic condition of the 918 mutation, thus proving to be a robust genotypic test for the knockout mutation.
Figure 8: Schematic representation of the RFLP procedure for the 4280 polymorphism using dCAP primers, which were used to introduce two SNPs based on the 4280 mutation. Creating two recognition sites, and subsequent digestion of the PCR products using specific endonucleases, HinfI and AflIII. Predicted bands of the RFLP products based on lengths of digested PCR fragments.
4280 polymorphism ambiguity
On sequence analysis of the 4280 polymorphism, no specific restriction enzymes were found to selectively differentiate the mutant and WT sequences (i.e., no restriction sites were present that differed between the two alleles). A modification of RFLP was created to acquire the desired polymorphic specificity, in which the PCR primers were redesigned (dCAPS FINDER 2.0®) to forcibly induce new polymorphisms based on the target 4280 mutation and thus creating recognition sites in the alleles (Figure 8).
A pilot study was conducted with amplification (see materials and methods) and incubation at 37°C for less than 24 hours in appropriate buffers containing 4 U/μl of HinfI. The digested fragments were run on a 2% polyacrylamide gel. However, unlike the 918 mutation, the 4280 polymorphism RFLP analysis proved relatively unyielding (data not shown) possibly due to a combination of factors, including primer inefficiency, non-specific endonuclease activity, mismatch intolerance, or background mutations in the 3’ end primer region. A dominant and a recessive sample showed clear band patterns (data not shown) suggesting the amplification and the digestion were successful. However, the same could not be said for the other samples. The primer inefficiency and mismatch intolerance could be greatly reduced through reducing annealing temperatures, since base pair specificity is a direct function of temperature. As for non-specific enzymatic activity, the period of incubation can be reduced given the above modifications prove ineffective.
Pea as a model
Pea was chosen for this study in order to exploit our extensive knowledge of auxin physiology in the species. Moreover, Pisum sativum was favoured as it is a diploid species hence simplifying genetics (inheritance) and possibly reducing gene redundancy. Peas are naturally self-fertilising with a tolerance of induced cross-pollination thus back-crossing (crossing to an un-mutagenized parental line), often used in mutagenesis procedures, is an available option. Genome sequencing for Pisum sativum is in progress, and the full sequence information of the closely-related model legume Medicago truncatula is publicly available to aid in comparative plant genomics and reverse genetic studies, such as our own.
Several factors have to be considered before initiating a TILLING program for genes of interest. As EMS only affects guanine (G) or cytosine (C) bases a significant proportion of the genome remains unmutated. The point mutations do not necessarily lead to noticeable changes in gene function as SNPs may induce silent mutations, which are characterised by no change in the substituted amino acid due to redundancy in the genetic code. The SNPs of interest are the ones that lead to missense (change in the substituted amino acid) and knockout mutations (encoding a premature stop codon). These mutations are likely to alter the function of the encoded protein. Of the latter two, knockout mutations are highly desirable, especially in the 5’ end of the open reading frame as the encoded protein is likely to be significantly impaired in function (truncated). Thus, any hypomorphic (reduced gene activity) or amorphic (arrested gene function) property might be associated with the function of the gene.
The genetic code of higher organisms are robust in nature that minimises the effects of EMS mutagenesis, hence it is beneficial to target conserved parts of the gene (exons). Mutations in these regions are likely to result in altered protein activity as they encode the domains that likely makeup the catalytic sites of the protein [9]. Taking these factors and the sequence analysis of conserved regions into account (Figure 3) (aligned with Medicago truncatula-data not shown) the PsTAR2 mutants with desirable polymorphisms (missense and nonsense mutations) were chosen in this study.
Gene redundancy
Peas are innately diploid in gene condition, however there is considerable evidence to suggest an ancient triplication event (paleopolyploidy) in the lineage (Papilionoideae ), thus increasing the number of homologous genes to three [10]. These genes might share functional redundancy or may have evolved subfunctionalisation, in which case the genes would be non-redundant. Unsurprisingly, two more inferred homologs of PsTAR2 were recently cloned and conditionally named PsTAR Mt 5 g 80, and PsTAR Mt 5 g 90. A high degree of functional redundancy might occur in the PsTAR gene family, possibly necessitating the production of double or triple mutants in order to sufficiently reduce functional activity. This can be further complicated if the mutant loci are tightly linked, thus impeding the production of multiple mutants. Alternatively, methods such as miRNA and RNAi can be deployed to target functionally redundant genes by designing partially specific RNA molecules. But these methods are confined to producing hypomorphic traits, which do not aid in studying gene interactions in a pathway.
Linking mutation to phenotype
The linking of phenotypic traits to genotype is often complicated in a reverse genetic approach that induces background mutations. Measuring auxin levels would be a first instinct when working with auxin biosynthetic mutants but this was delayed due to visible mutant traits in the generations caused by non-specific background. Thus, no deficiency or trait can be conclusively attributed to the gene condition until a clear background is created. However, as an alternative to lengthy back-crossing programs certain correlations can be drawn from traits that are segregating in the same populations that are uniform in the background mutations they carry i.e., plants that are homozygous dominants, heterozygotes, and homozygous recessives of the same mutant family (e.g., 4280/4) carry essentially the same background. Thus, any traits observed in the recessive condition, relative to the dominant and heterozygote can be attributed with certainty to the perturbed gene.
In conclusion, the mutation for colour (cc) was found to be associated with PsTAR , possibly denoting closely linked loci or less likely an effect of the PsTAR2 mutation. The 4280 mutation has at most a subtle effect on plant phenotype. The highly desirable IPyA knockout mutation (918) has been successfully isolated and will effectively aid future studies involving IPyA auxin-biology. Together the 4280 and 918 mutants constitute an allelic series that range in functional activity and will effectively aid future studies involving complex gene functions and interactions that interplay in the IPyA pathway.
The advent of TILLING has greatly benefited reverse genetics in model species such as peas, which are innately resistant towards conventional transformation (Agrobacterium). The procedure is costeffective and coupled with high-throughput screening it is proving to be the best currently available option in targeted mutagenesis. The frequent generation of missense polymorphisms by the procedure that leads to an allelic series of mutants is of great importance to geneticists who want to study gene functions and interactions. The benefits of the procedure are immense but there are a few drawbacks involved, including high labour intensity and non-specific background. However, the pros outweigh the cons and for many researchers working on non-transformable model species, such as peas, there are few reverse genetic alternatives to TILLING.
I the author would like to thank Sree Balaji Medical College and Hospital and its associated labs for their help and support towards this study.
The author declares that no conflict of interest exists.