Journal of Developing Drugs
Open Access
ISSN: 2329-6631
ISSN: 2329-6631
Research Article - (2022)Volume 11, Issue 4
The present work contains details of formulating and evaluation of mouth dissolving tablets of micronized terbutaline sulphate by using co-processed superdisintegrants and taste masking agent (flavors and ion exchange resin) with an aim to get a dosage form having fast disintegration.
Terbutaline sulphate first pass through size reduction technique which leads to increase in surface area and increased in oral absorption of the drug. As a part of pre-formulation study, the physicochemical compatibility study of the drug with different excipients was done and drug-excipient interaction was assessed from thermal analysis by using differential scanning calorimetry indicated absence of interactions between the selected excipients for the study. Moisture absorption study of the drug indicated that the drug is non-hygroscopic. Thermal (stress) Stability of the drug was also assessed and found to be satisfactory.
Factorial Design of research methodology was used for formulation design study by keeping consideration for variable ingredients which directly affect the disintegration and palatability of the formulation. Direct compression method was employed in the manufacturing of tablets with different superdisintegrants and flavours and their concentrations, but two formulations were also designed using ion exchange complex method for masking the taste by wet granulation method.
The selected formulations (four) were subjected for complete (detailed) evaluation of critical quality attributes like uniformity of weight, hardness, thickness, friability, disintegration time, wetting time (water uptake (swelling) and erosion studies), drug content and dissolution profile.
Superdisintegrants; Disintegration; Hardness test; Flavors
The buccal region of mouth offers significant route of administration for systemic drug delivery. The mucosa has a rich supply of blood and it is relatively permeable. Oral route is most preferred to the patient and the clinician alike. However, peroral administration of drugs has disadvantages such as hepatic first pass metabolism enzymatic degradation within the GI tract that prohibits oral administration. Oral cavity route of delivery offers distinct advantages including possible by pass of first pass effect, avoidance of pre systemic elimination within the GI tract.
It has been known for centuries that buccal and sublingual administration of drugs results in rapid absorption into the reticulated vein, which lies underneath the oral mucosa and transport through the facial veins, internal jugular vein, and brachiocephalic vein into the systemic circulation. Therefore, the buccal and sublingual routes of administration can be utilized to bypass the hepatic first-pass elimination of drugs. In the oral mucosal cavity, the buccal region offers an attractive route of administration for systemic drug delivery. The mucosa has a rich blood supply and it is relatively permeable [1].
The CDER nomenclature standards committee states the below mentioned definition for an ODT as a new dosage form in 1998. A solid dosage form containing medicinal substances which disintegrates rapidly, usually within a matter of seconds, when placed upon the tongue.
Characteristics that were exhibited by the initial products included low tablet weight, small tablet size, highly soluble components, and rapid disintegration. European pharmacopoeia has used the term orodispersible tablets for tablets that disperse inside the mouth immediately and within 3.0 minutes before swallowing. As tablet disintegrates in mouth it enhances the clinical effect of drug through pre-gastric absorption from mouth pharynx and esophagus. These are novel type of tablets that disintegrate/dissolve/disperse in saliva within few seconds. The basic approach used in development of mouth dissolving tablet is the use of superdisintegrants like cross linked carboxymethylcellulose (crosscarmellose), sodium starch glycolate (primogel, explotab), polyvinylpyrrolidone (polyplasdone) which provide instantaneous disintegration of tablet in mouth thereby releasing the drug in saliva [2].
Orally disintegrating tablets are an appealing dosage form for many reasons. Health professionals find the mouth disintegrating tablets as a good alternative for traditional tablets and liquid forms. Pediatric, geriatric, bedridden, and developmentally disabled patients are especially well suited for this alternative to traditional tablets. Medications used for treating nausea, allergies, migraines, arthritis, depression, and schizophrenia are already available as mouth dissolving tablets form.
Asthma is the most common chronic illness of childhood affecting approximately 10% of children. Persistent asthma is managed pharmacologically by the daily use of a controlled medication and a short acting beta-agonist inhaler for relief of exacerbations. Inhaled corticosteroids are often used as daily controller therapy for patients with persistent asthma. However, long term inhaled corticosteroids exhibit dose related systemic side effects. Moderate to high doses have been associated with a transiently decreased rate of growth in children, decreased bone mineral density, and the development of glaucoma in adults. Inhaled cromolyn or nedocromil have a better safety profile and may be considered in children, but patients with asthma especially children are often poorly adherent to these frequently inhaled therapies and have problems to achieve adequate delivery. The commonly used drugs in asthma are adrenaline, ephedrine, isoprenaline, salbutamol and terbutaline [3].
The extent of the bioavailability of terbutaline sulphate after oral administration is 7% to 26%. This decrease in the percentage is due to a high first-pass metabolism. The terminal half-life in healthy subjects is 17 hours and the biological half-life is 3.6 hours. Terbutaline is a β2-receptor agonist similar to epinephrine. Nevertheless, a change from a catechol-like structure to a resorcinol-like structure and the use of a bulky amino substituent make it β2 selective, unlike epinephrine.
Incomplete absorption from the gastrointestinal track and a fairly large first-pass metabolism make terbutaline a suitable candidate for orally disintegrating tablets.
Materials
Terbutaline sulphate (API) was obtained as a gift sample from Unicure India Limited, NOIDA and HiGlance Laboratories, Greater Noida. All Excipients i.e., mannitol, lactose (Pharmatose 200 M), aspartame, sodium starch glycolate, Ac-Di-Sol, PPXL-10, magnesium stearate, talc, and favours, orange DC, menthol and mint) were provided as a gift sample from Modi-Mundipharma R&D Centre, Modipuram, and Meerut.
Micronization of terbutaline sulphate: Terbutaline sulphate first pass though the sieve to make it irregular in shape Microscopic study of Terbutaline sulphate was carried out with 40X microscopic magnification (Camera make: Lucida) which reveals that molecules in terbutaline sulphate is arranged in polymorphic fashion.
Microscopic view confirms that the drug is micronized grade irregular shape crystalline material and suitable for direct compression (Figure 1). Micronized particles will provide larger surface area which will enable higher solubility as larger surface area allows a greater interaction with the solvent and this leads to a better dissolution profile. Hence drug is suitable for mouth dissolving tablets [4].

Figure 1: Microscopic view of terbutaline sulphate.
Selection of process
The concept of wet granulation cannot be used for designing the mouth dissolving tablets as this type of process involves waterbased binder which may leads to high disintegration time of a tablet. The use of slugging process was also not considered because it is a cumbersome process [5].
Since the drug product involves low strength of API It has been decided to select the direct compression method which does not involve water or water-based binders and requires simple mixing process only Mouth dissolving tablets were prepared by direct compression method as per the Table1 two more formulations were designed.
| Batch Number | B1 | B2 | B3 | B4 | B5 | B6 | B7 | B8 | |
|---|---|---|---|---|---|---|---|---|---|
| Ingredients | mg/tab | mg/tab | mg/tab | mg/tab | mg/tab | mg/tab | mg/tab | mg/tab | |
| Terbutaline Sulphate | 5.00 | 5.00 | 5.00 | 5.00 | 5.00 | 5.00 | 5.00 | 5.00 | |
| Aspartame | 0.20 | 0.20 | 0.20 | 0.20 | 0.20 | 0.20 | 0.20 | 0.20 | |
| Mannitol | 60.00 | 60.00 | 60.00 | 60.00 | 60.00 | 60.00 | 60.00 | 60.00 | |
| Magnesium stearate | 0.50 | 0.50 | 0.50 | 0.50 | 0.50 | 0.50 | 0.50 | 0.50 | |
| Talc | 0.50 | 0.50 | 0.50 | 0.50 | 0.50 | 0.50 | 0.50 | 0.50 | |
| Lactose | 25.80 | 21.80 | 17.80 | 25.80 | 17.80 | 25.80 | 21.80 | 33.80 | |
| Superdisintegrants | |||||||||
| SSG | 4.00 | 4.00 | 4.00 | ||||||
| Ac-Di-Sol | 4.00 | 4.00 | 4.00 | 4.00 | 4.00 | ||||
| PPXL 10 | 4.00 | 4.00 | 4.00 | 4.00 | |||||
| Flavors | |||||||||
| Menthol | 4.00 | 4.00 | 2.00 | ||||||
| Orange DC 100 | 4.00 | 4.00 | 2.00 | 2.00 | |||||
| Mint | 4.00 | 4.00 | 2.00 | ||||||
Table 1: Formulation and disintegration methods of tablets.
Formulation no. i.e. B5 and B7 were selected as a basic formulation, based on good palatability and better disintegration time out of all prototype formulations, for further improvement [6]. Further, their DT was improved by increasing the concentration of polyplasdonecrospovidone. The new formulations were termed as B5A and B7A (Table 2).
| Batch Number | B5A | B7A |
|---|---|---|
| Ingredients | mg/tab | mg/tab |
| Terbutaline sulphate | 5.00 | 5.00 |
| Aspartame | 0.20 | 0.20 |
| Mannitol | 56.00 | 56.00 |
| Mag stearate | 0.50 | 0.50 |
| Talc | 0.50 | 0.50 |
| Lactose | 17.80 | 21.80 |
| Superdisintegrants | ||
| SSG | - | - |
| Ac-Di-Sol | 4.00 | 4.00 |
| PPXL 10 | 8.00 | 8.00 |
| Flavors | - | - |
| Menthol | 2.00 | |
| Orange DC 100 | 2.00 | 2.00 |
| Mint | 4.00 | 2.00 |
Table 2: Additional formulations with high concentration of PPXL 10.
Taste masking with ion exchange complexation: A latest taste masking agent i.e. Kyron T 114 (Manufactured by Corel Pharma) was used in formulation no. B5/B7 to improve the palatability of of drug product [7]. The new formulations were termed as B9 and B10 which were similar in ingredients and their concentration but different in processing for taste masking (Table 3).
| Batch Number | B9 | B10 |
|---|---|---|
| Ingredients | mg/tab | mg/tab |
| Terbutaline Sulphate | 5.00 | 5.00 |
| Aspartame | 0.20 | 0.20 |
| Mannitol | 59.00 | 59.00 |
| Mag stearate | 1.00 | 1.00 |
| Talc | 1.00 | 1.00 |
| Lactose | 14.8 | 14.8 |
| Taste Masking Agent | ||
| Kyron T 114 | 5.00 | 5.00 |
| Superdisintegrants | ||
| SSG | - | - |
| Ac-Di-Sol | 4.00 | 4.00 |
| PPXL 10 | 4.00 | 4.00 |
| Flavors | ||
| Menthol | 2.00 | 2.00 |
| Orange DC 100 | ||
| Mint | 4.00 | 4.00 |
Table 3: Drug product for taste masking process.
Micrometric properties of the powder blend: Before compression powder blend was evaluated for the bulk density, tapped density, angle of repose, carr’s index, hausner ratio. Bd and Td was determined by tapped density apparatus (Electrolab) apparatus, angle of repose was determine by fixed funnel method by placing ten gram of powder blend in a plugged glass funnel and then allow to flow through funnel orifice4. Results are shown in Table 4.
| Micrometric Properties | Results |
|---|---|
| Bulk density (Bd) | 0.600 to 0.667 gm/ml |
| Tapped density (Td) | 0.714 to 0.789 gm/ml |
| Angle of repose | 19.95 to 21.28° |
| Carr’s index (CI) | 14.89 to 16.00% |
| Hausner’s Ratio (HR) | 1.175 to 1.190 |
Table 4: Micrometric properties of the powder blend.
Direct compression process parameters: Compression of all the batches was carried out by using single rotary 8 station compression machine (Make: Cadmach). The compression was carried out considering parameters and specifications as per (Tables 5 and 6).
| Lower and upper punch specification | 6.5 mm and round concave |
|---|---|
| Standard. weight of 10 tablets | 1.00 g ± 5.0% |
| Individual weight variation weight per tablet | 100.00 mg ± 7.5% |
| Hardness | NLT 3 KP |
| Thickness | Only observation |
| Friability | NMT 1.0% w/w |
| Satisfactory appearance | Off-white cores free from imperfections i.e., no appearance defect like sticking, picking or rough surface |
Table 5: Compression process parameters and specifications.
| Batch number | B1 | B2 | B3 | B4 | B5 | B6 | B7 | B8 | B5A | B7A | B9 | B10 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Av. Weight (mg) | 100.20 | 100.10 | 100.50 | 100.15 | 100.20 | 100.28 | 100.10 | 100.15 | 100.05 | 100.10 | 100.25 | 100.30 |
| Hardness (KP) | 2.l7 -3.20 | 2.60 - 3.70 | 6.30 - 9.20 | 7.00 - 9.00 | 3.50 - 4.50 | 3.00 - 4.00 | 4.00 - 5.50 | 4.00 - 5.50 | 4.0 – 5.0 | 4.0-5.0 | 4.0 - 5.0 | 4.0 - 5.0 |
| Thickness (mm) | 2.85 -2.95 | 2.90 - 3.00 | 2.95 -3.05 | 2.95 - 3.00 | 2.95 -3.05 | 2.95 -3.05 | 2.90 - 3.00 | 2.95-3.05 | 3.0- 3.1 | 3.0-3.1 | 3.0 - 3.1 | 3.0 - 3.1 |
| DT (Seconds) | 70 | 60 | 45 | 90 | 70 | 50 | 60 | 80 | 24 | 16 | 180 | 30 |
| Palatability | + | + | ++ | ++ | ++ | +++ | ++ | +++ | +++ | ++ | ++ |
Table 6: The results of compressed tablets for physical parameters.
Evaluation of Terbutaline sulphate MDT: Formulation of selected batches i.e. B.No. B3, B5A, B7A and B10 were subjected to following quality control tests.
General appearance
White, round and biconvex uncoated tablets. Diameter of all the tablets was between 6.5 ± 0.2 mm the tablets were free from imperfections like sticking/picking or rough surface (Figure 2).

Figure 2: White, round and biconvex uncoted tablets.
Uniformity of weight: Test for Uniformity of weight (weight variation) was carried out as per Indian pharmacopoeia [8]. Twenty (20) tablets were taken and their weight was determined individually and collectively on a digital weighing balance (Make: Mettler Toledo) as per Table 7.
| Formulation | Average weight (mg) | Minimum weight (mg) | Maximum weight (mg) | Variation (%) |
|---|---|---|---|---|
| B3 | 102.1 | 99 | 104 | -1 to +4 |
| B5A | 101.8 | 99 | 104 | -1 to +4 |
| B7A | 102.4 | 99 | 105 | -1 to +5 |
| B10 | 102 | 99 | 105 | -1 to +5 |
Table 7: Uniformity weight of tablets as per Indian pharmacopeia.
Disintegration time: The test was carried out on the 6 tablets using the DT apparatus (Make: Electrolab) as specified in IP using purified water at 37°C ± 2°C as a disintegration medium and the time taken for complete disintegration of the tablet with no palpable mass remaining in the apparatus was measured in seconds as per Table 8.
| S.No. | Formulation | Disintegration Time (Sec) |
|---|---|---|
| 1 | B3 | 48 |
| 2 | B5A | 21 |
| 3 | B7A | 17 |
| 4 | B10 | 30 |
Table 8: Disintegration of the tablet friability.
Friability: Friability test apparatus (Make: Electrolab, Mumbai) was used to check the friability of the selected formulations. A weight of not less than 6.5 gm was revolved at 25 rpm for 04 min with a dropping distance of 6 inch with each revolution. After completion of rotations for 4 min. results of friability was as per Table 8.
Hardness: The aim of Hardness Test is for determination of tablet breaking force. This is the force required to crush a tablet by applying across the diameter of the tablet. Hardness of each formulation was determined by using Electro lab Hardness Tester (EHT-5PR) by using 6 tablets from each selected formulation and evaluated and results were as per Table 9.
| S.No. | Formulation | Average hardness (KP) |
|---|---|---|
| 1 | B3 | 3.2 |
| 2 | B5A | 3.4 |
| 3 | B7A | 3.3 |
| 4 | B10 | 3.3 |
Table 9: Cumulative results for hardness test.
Thickness: Thickness test of each formulation was carried out by using Electro lab Hardness Tester (EHT-5PR) by using 6 tablets from each selected formulation and evaluated. Individual as well as average value was used interpretation of results as per Table 10.
| S.No. | Formulation | Average thickness in mm |
|---|---|---|
| 1 | B3 | 3.14 |
| 2 | B5A | 3.13 |
| 3 | B7A | 3.13 |
| 4 | B10 | 3.08 |
Table 10: Cumulative results for Thickness test.
Wetting time: Wetting time is considered as an important criterion for determining the capacity of disintegrating agent to swell in presence of little amount of water. A piece of tissue paper, folded twice, was placed in a small petridish (internal diameter =9.0 cm) containing 9.0 ml of distilled water and water-soluble dye i.e. methylene blue. A tablet was kept on the paper, and the time for complete wetting was measured. Results were shown in Table 11.
| S.No. | Formulation | Average Wetting time (in sec) |
|---|---|---|
| 1 | B3 | 58 |
| 2 | B5A | 45 |
| 3 | B7A | 37 |
| 4 | B10 | 37 |
Table 11: Cumulative results for wetting time.
In-Vitro dissolution studies
In vitro dissolution studies for the tablets was carried out by USP XXIV paddle method at 100 rpm in 900 ml of water as dissolution media, maintained at 37°C ± 0.5°C. 5.0 ml of the medium was withdrawn at the specified time intervals, filtered and absorbance was checked by using UV-visible spectrophotometer.
An equal volume of fresh medium, prewarmed at 37°C was added to the dissolution medium after each sampling to maintain the constant volume throughout the test (Table 12).
| S.No. | Formulation | % Drug release | ||
|---|---|---|---|---|
| 2.5 min | 2.5 min | 2.5 min | ||
| 1 | B3 | 93.3 | 101.9 | 102.2 |
| 2 | B5A | 92.1 | 99.7 | 100.6 |
| 3 | B7A | 93.2 | 101.4 | 101.9 |
| 4 | B10 | 92 | 101.9 | 102.8 |
Table 12: Dissolution results for drug release.
Sampling Profile: 2.5, 5, 15, 30 and 45 minutes, further sampling was stopped when drug release was observed above 90.0%. Results of selected formulation shown in Figure 3.

Figure 3: Drug release v.s time plot. Note: B3,
B3,  B5A,
B5A,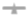 B7A,
B7A,  B10.
B10.
Terbutaline sulphate tablets were prepared by direct compression method. Accurately weight amount of API and Excipients are taken. The micrometric properties of powder blend of drug and excipients are shown in Table 3 all parameters were found within the limit and do not affect the compression of the tablets. The compressed tablets were evaluated, average weight of all formulation were found between 100.05 to 100.50 mg. Tablet breaking force (hardness) was found between 2.17 to 3.10 kP, thickness of all formulation were found between 2.85 to 3.10, Disintegration time of all formulations were found between 16 to 120 sec on the basis of compression data further 04 formulation i.e., B3, B5A, B7A and B10 were selected.
From the present investigation, it may conclude that mouth dissolving tablets of terbutaline sulphate can be formulated by direct compression method by using micronized terbutaline sulphate with co-processed superdisintegrants. The present study reports successful formulation of mouth dissolving tablet of terbutaline sulphate. Direct compression method was employed in the manufacturing of tablets with different superdisintegrants and flavours and their concentrations, but two formulations were also designed using ion exchange complex method for masking the taste by wet granulation method.
[Google scholar] [Crossref] [Pubmed]
[Google scholar] [Crossref] [Pubmed]
Citation: Sharma D, Aggarwal R, Tomar R (2022) Formulation and Evaluation of Mouth Dissolving Tablets Prepared by Micronized Terbutaline Sulfate and Using Co-processed Superdisintegrants by Direct Compression Method. J Develop Drugs 11:179.
Received: 04-Jul-2022, Manuscript No. EOED-22-001-PreQC-22; Editor assigned: 08-Jul-2022, Pre QC No. EOED-22-001-PreQC-22 (PQ); Reviewed: 22-Jul-2022, QC No. EOED-22-001-PreQC-22; Revised: 29-Jul-2022, Manuscript No. EOED-22-001-PreQC-22 (R); Published: 05-Aug-2022 , DOI: : 10.35248/2329-6631.22.11.179
Copyright: © 2022 Sharma D, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.