Journal of Theoretical & Computational Science
Open Access
ISSN: 2376-130X
ISSN: 2376-130X
Research Article - (2016) Volume 3, Issue 1
In this work we investigate the copolymerization process of 1,3-butadiene with vinyl chloride by means of density functional theory and transition state theory. The purpose of this study is to determine how reliable a first principles approach can predict the copolymer composition curve. Previous studies have shown that it is dificult to obtain quantitatively accurate rate constants from first principles for polymerization processes. However, since the copolymerization process depends on the relative rates of competing mechanisms a possible cancellation of errors can improve the predictability of this method. Using a moderate level of theory and basis set we obtain qualitatively accurate predictions of the copolymer composition curve.
<Keywords: Density functional theory; Transition state theory; Copolymerization/polymerization; Kinetic modelling; Quantum Mechanics
Chemical engineers rely upon the accuracy of kinetic models when developing polymerization processes. Typically, these models are developed with a top-down approach, namely using macroscopic experimental data to deduce molecular kinetic phenomena. Common examples of experimental `observables’ include the overall rate of reaction, molecular weight distributions, and the concentration of certain species with respect to time. Due to the inherent complexity of radical polymerization reactions, model based assumptions are necessary to simplify the reaction network. These assumptions reduce the number of rate parameters (such as the pre-exponential factor and the activation energy (Ea)) that are included in the regression scheme of the experimental data. Historically, it has been the job of the chemist to propose the most feasible reaction mechanisms in order to relate the macroscopic data to the molecular rate constants. Excluding and/or combining certain reactions introduce a source of error that is di cult to quantify. This brings into question the reliability of the model when used at conditions far removed from those of the experimental data [1].
An alternative approach that has gained much attention in the last decade is known as ab initio kinetic modeling [2]. Rather than taking a top-down approach, this method starts at the molecular level without relying on any experimental data. One clear advantage of this approach is that expensive and time-consuming experiments are not required. This allows the engineer to predict whether or not new processes will be profitable a priori. Furthermore, because the rate constants are not t to experimental data, no assumptions need to be made to simplify the reaction mechanism. That is to say that all reaction pathways can be included if desired. It is common to exclude certain pathways that have large activation barriers but this is based upon first principles knowledge of the rate constants, rather than a chemist’s intuition [3]. Another advantage of the ab initio approach is that it provides deeper insight on a molecular level into why certain phenomena are observed on a macroscopic level. That being said, in most cases to date the extent of that insight has been qualitative. These qualitative results include: the geometry of the transition state (TS) [4,5], which reaction mechanisms are most likely [6,7], and whether penultimate effects are negligible [8]. Only a hand full of polymer studies has reported quantitatively accurate results [9]. The validation of the quality of these results is often a comparison between macroscopic experimental data and the experimental data predicted from the ab initio model [10]. However, obtaining accurate quantitative results requires computationally intensive calculations [1].
Copolymerization is an industrially relevant example where an accurate kinetic model is essential. This process can produce polymers that have a combination of physical properties that are unattainable from a pure polymer. The physical properties of the resultant copolymer are strongly dependent on whether it has a random, block, graft, or alternating configuration. The reaction conditions and relative reactivity of the monomers dictate the type of configuration as well as the composition of the copolymer. The copolymer composition can be described by the integrated copolymer equation.
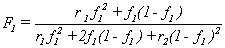 (1)
(1)
where F1 is the composition of the copolymer produced, f1 is the composition of the monomer ‘pool’, and r1 and r2 are the reactivity ratios for monomer 1 and 2, respectively. An underlying assumption in the derivation of this equation is that penultimate effects are negligible. This means that only the identity of the terminal species of a radical determines its reactivity. With this assumption, in order to predict the copolymer composition for a known monomer ‘pool’ composition, all that is needed are the rate constants for the four different monomer addition reactions [11].
In this study, we have chosen a relatively simply copolymerization process to model, namely, 1,3-butadiene (B) with vinyl chloride (C). The primary reason we selected these smaller monomers is to reduce the computational cost of the ab initio calculations. Another reason these compounds were selected is because experimental r1 and r2 values are available, which allows for quantitative assessment (where B is monomer 1 and C is monomer 2) [11]. Furthermore, r1 is much greater than one and r2 is much less than one, meaning those radicals prefers to react with monomer B. Whether or not this trend is observed from our calculations provides a qualitative measure for the validity of our results. The objective of this project is threefold: to demonstrate how ab initio calculations can be used to develop kinetic models, to verify the qualitative trend for relative reactivity’s, and to determine whether or not quantitative accuracy is obtainable for engineering design purposes.
The outline for this document is the following. In Section 2 we explain the theoretical basis for this work. A description of the methodology follows in Section 3. In Section 4 we present and analyse the results. We consider possible limitations of these results in Section 5. Finally, in Section 6 we summarize the main conclusions.
The geometry optimization of the reactants and products involves perturbing the bond lengths, angles, etc. and finding the lowest energy geometry. The energy is determined by a numerical approximation to the solution to the Schrodinger equation using the given basis set. These optimization methods are reliable for stable species that pertain to a minimum in the potential energy landscape. However, in order to predict rate constants from quantum mechanical calculations, the transition state must also be located. This ephemeral state cannot be measured experimentally but can be predicted with computational chemistry. The TS corresponds to a rst-order saddle-point on the energy landscape, meaning that it is a minimum in all reaction coordinates except for one, the intrinsic reaction coordinate. For our purposes, suffice it to say that locating the TS is a computationally demanding task that requires hours of rigorous optimization techniques. Having located the transition state, the rate constant (k) for a bimolecular reaction can be obtained from statistical mechanics.
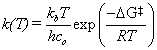 (2)
(2)
where kb is Boltzmann’s constant, T is the absolute temperature, h is Planck’s constant, co is the standard unit of concentration, 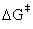 is the change in Gibbs free energy from reactants to TS, and R is the universal gas constant [3]. However, since the experimental data are for reactivity ratios (r), we can take the ratio of rate constants to arrive at
is the change in Gibbs free energy from reactants to TS, and R is the universal gas constant [3]. However, since the experimental data are for reactivity ratios (r), we can take the ratio of rate constants to arrive at
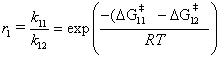 (3)
(3)
where the subscripts 11 and 12 refer to monomer 1 or 2, respectively, adding to a radical with a terminal monomer 1. A similar expression can be obtained for r2. Although accurate ab initio predictions of rate constants are difficult, we expect the reactivity ratio, r, to be more accurate because Equation 3 depends on the difference in values. In other words, the ab initio method is reliable as long as the error in is the same for the two reactions being compared. Since Equation 1 depends upon r1 and r2, even though the rate constants for each individual step may not be quantitatively accurate we hypothesize that the copolymer composition curve will be qualitatively, if not quantitatively, reliable. Testing this hypothesis consists of two of the three primary objectives of this work outlined in Section 1.
The method described previously is general and applies to all ab initio reaction modelling. However, polymerization modelling requires an additional step, namely providing a surrogate polymer. A surrogate polymer is a radical molecule that consists of a reduced number of monomers with an initiator connected to one end. This is necessary because computational costs increase dramatically with increasing chain-length [12]. The literature suggests that a backbone of four atoms is the minimum for convergence to the true polymer limit [2]. For this reason, in this work we have used a four atom chain-length as the minimum surrogate polymer size.
In this work, we utilize Gaussian 03 for all quantum mechanical calculations. The reaction scheme consists of four different reactions; two reactions with B as the terminal monomer and two with C as the terminal monomer. The two different radicals undergo two independent propagation reactions by addition of a B or C monomer. The initiator group (I) is found at the beginning of each species and, for computational purposes, I is usually just a CH3 group. With this definition for I, however, IC would not meet the required backbone chain-length of four atoms. For this reason, originally we chose IBC for reactions 3 and 4 (see below). However, locating the transition state for reaction 4 (TS4) is significantly more challenging than the other transition states. Therefore, we investigate whether or not a smaller surrogate can properly model reaction 4 by changing I to a C3H5 group in reaction 5. The five reactions studied in this work are.
IB + B → IBB (R1)
IB + C → IBC (R2)
IBC + C → IBCC (R3)
IBC + B → IBCB (R4)
IC + B → ICB (R5)
These reactions consist of nine unique reactants and products and five transition states. The shorthand notation for the nine stable species and their chemical structures are displayed in Figure 1. In order to assess the need for a larger basis set, we use two different methods for calculating the energies. As recommended by the literature, the optimal geometries are all located using a computationally cheap B3LYP/6-31G(d) model. However, a significantly more expensive method is employed that is known to improve energy calculations by a factor of three [13]. This method, B3LYP/6-311+G(3df,2p)//B3LYP/6- 31G(d), is a well-established composite method that uses the same geometries obtained previously but utilizes a larger basis set for the energy calculation. Although the literature recommends the G2-(MP2) composite method, this method only provides marginal improvement while requiring significantly more computational time [14]. Also, it has been shown previously that the activation barrier does not converge to a single value with higher levels of theory and increasing basis sets. In fact, in some cases these lower levels of theory and small basis sets yield results that are almost identical to much more demanding methods (see Table 2 in Ref [14]). Furthermore, because we compare the ratio of reaction rates rather than the reaction rate itself, we assume that our results do not differ much from a more computationally demanding approach, due to a cancellation of errors in the values.

Figure 1: Shorthand notation for reactants and products used in this study. The color scheme is light gray, dark gray, and green corresponding to Hydrogen, Carbon, and Chlorine, respectively.
In this work, no special methods are utilized to verify that a global minimum is obtained from the geometry optimizations. Although conformation search algorithms exist, they require inordinate amounts of computational time for molecules of the size used here. Chemical intuition is usually sufficient to verify that the conformation looks appropriate. To assure that the correct TS is located; we verify that the imaginary frequency corresponds with the proper intrinsic reaction coordinate [13].
Furthermore, a single level of theory and basis set are used for the entire molecule, rather than utilizing a layered approach, such as ONIOM. Layered methods apply a higher level of theory and larger basis set to the region where the reaction takes place (the radical and approaching double bond) while a simpler method is used for the rest of the molecule. This method is effective because most of the molecule is unaffected by the polymerization process. However, these methods are still more complicated than the simple B3LYP/6-31G(d) and B3LYP/6- 311+G(3df,2p)//B3LYP/6-31G(d) methods used here [9].
The optimal geometries for the reactants and products are found in Figure 1. The geometries obtained for each TS are displayed in Figure 2. It is important to note that the pathway of vinyl chloride addition is quite different depending on if the radical has a terminal B or C (compare TS2 to TS3). Also, it is reassuring that the geometries in TS4 and TS5 look very similar despite using two different surrogate sizes. This is significant because the time required to locate TS5 is an order of magnitude smaller than that for TS4.

Figure 2: Geometries of the transition states for each reaction. All geometries were optimized with B3LYP/6-31G(d). The color scheme is light gray, dark gray, and green corresponding to Hydrogen, Carbon, and Chlorine, respectively.
Table 1 contains the energies for each species calculated with the two different methods. The zero-point energies (ZPE) are scaled with the appropriate factor for the B3LYP/6-31G(d) vibrational calculations. Notice that the energies obtained from the larger basis set are more negative than those calculated with the smaller basis set. This is consistent with the variation principle and suggests that the larger basis set results are more accurate [13].
| 6-31G(d) | 6-311+G(3df,2p) | ||||
|---|---|---|---|---|---|
| Species | ZPE | E0 | G2980 | E0 | G2980 |
| B | 0.08549 | -155.99214 | -155.93475 | -156.05192 | -155.99453 |
| C | 0.04286 | -538.18539 | -538.16882 | -538.25066 | -538.23409 |
| IB | 0.12328 | -195.89208 | -195.80038 | -195.96338 | -195.87167 |
| IC | 0.11501 | -655.46553 | -655.38629 | -655.57017 | -655.49094 |
| IBC | 0.17206 | -734.09867 | -733.96825 | -734.22822 | -734.09780 |
| IBB | 0.21442 | -351.91793 | -351.74639 | -352.04143 | -351.86989 |
| ICB | 0.20545 | -811.50972 | -811.34893 | -811.66643 | -811.50563 |
| IBCC | 0.22050 | -1272.32184 | -1272.14993 | -1272.50957 | -1272.33767 |
| IBCB | 0.26249 | -890.14281 | -889.93079 | -890.32440 | -890.11239 |
| TS1 | 0.21094 | -351.8737 | -351.70635 | -352.00091 | -351.83356 |
| TS2 | 0.16868 | -734.06380 | -733.93692 | -734.19641 | -734.06953 |
| TS3 | 0.21660 | -1272.27482 | -1272.10852 | -1272.46658 | -1272.30029 |
| TS4 | 0.25908 | -890.08530 | -889.87861 | -890.27157 | -890.06489 |
| TS5 | 0.20194 | -811.4521646 | -811.29711 | -811.6135377 | -811.45848 |
Table 1: Zero-Point Energy (ZPE), Electronic Energy (E0), and Standard Gibbs Free Energy at 298 K (G0298) of optimized geometries for both basis sets (all values are in Hartrees).
A comparison between the results from the two different basis sets provides some valuable insight. Table 2 presents the activation energies (Ea) and the change in electronic energy (ΔE0) for each reaction. Notice that the higher accuracy energy calculations predict larger activation barriers and less exothermic reactions. This means that a smaller basis set would favour a more reactive polymerization process both on a kinetic and thermodynamic basis. Furthermore, notice that Ea and ΔE0 for reaction 4 and 5 are nearly identical. This suggests that the smaller surrogate used in reaction 5 is sufficient, especially considering the uncertainty associated with each value. In order to validate our results, Table 3 provides a comparison between the reactivity ratios obtained in this study with those available in the literature. It is interesting to note that the smaller basis set actually resulted in a r1 value that agrees marginally better with the experimental value. Although it was seen in Table 2 that the difference between the results from reaction 4 and 5 was small, the smaller surrogate does exhibit a stronger deviation from the literature. That being said, a key observation from Table 3 is that the correct qualitative trend is observed, namely, that r1 is much greater than one while r2 is much less than one. This means that our ab initio approach successfully predicts that addition of 1,3-butadiene is strongly preferred regardless of which monomer is the terminal species. Although the reactivity ratios differ from the experimental values by over 100%, from an engineering standpoint the real concern is how much this will affect the design of the plant. This can best be observed by plotting the polymer composition with respect to the monomer ‘pool’ composition as predicted by the copolymer equation (Equation 1). As seen in Figure 3, the different ab initio methods are indiscernible. This means that the smaller surrogate and smaller basis set predict a nearly identical process design to that of the computationally expensive larger surrogate and larger basis set. Furthermore, the ab initio kinetic model accurately predicts the qualitative nature of the copolymer trend. That being said, the deviation from the experimental trend is significant and suggests that this method is not adequate for quantitative purposes. Since we observed only a small change with respect to the size of the basis set and surrogate chain-length, we attribute this error to the geometries obtained for the transition states. Specifically, the TS geometry results may be more sensitive to the level of theory used than the reactants and products because the TS is a delicate saddle-point on the energy landscape. Future work would require an investigation of the transition state structures at a higher level of theory or with a more sophisticated ONIOM method.
| 6-31G(d) | 6-311+G(3df,2p) | |||
|---|---|---|---|---|
| Reaction | Ea | E0 | Ea | E0 |
| 1 | 7.94 | -17.68 | 10.36 | -12.93 |
| 2 | 10.15 | -9.65 | 12.63 | -5.25 |
| 3 | 6.84 | -20.27 | 8.75 | -15.83 |
| 4 | 4.40 | -29.59 | 6.31 | -24.74 |
| 5 | 4.34 | -29.62 | 6.25 | -24.78 |
Table 2: Comparison of reaction energies for both basis sets (all values are in kcal/mol).
| Reactivity Ratios | Exp. | 6-31G(d) | 6-311+G(3df,2p) |
|---|---|---|---|
| r1 | 8.8 | 30.57 | 33.41 |
| r2 (with TS4) | 0.035 | 0.0171 | 0.0171 |
| r2 (with TS5) | 0.035 | 0.011 | 0.011 |
Table 3: Comparison of experimental reactivity ratios [11] with those obtained from the different ab initio methods. Values are reported at 323.15 K.

Figure 3: A comparison between the experimental data and the four different ab initio results. The curves were generated using the copolymer equation with the values found in Table 3. The compositions are in terms of 1,3-butadiene.
For simplicity, we have assumed that penultimate effects are negligible. Specifically, we assumed that the penultimate group was a hydrocarbon, typically the 1,3-butadiene group. This assumption was justified by the fact that the vinyl chloride group adds to the growing polymer much less frequently than 1,3-butadiene. However, the validation of this assumption is left as future work.
The system we investigated is a very simple copolymerization process because r1 >>> 1, r2 <<< 1, and r1r2 ≠ 1. More difficult systems to obtain qualitatively accurate results are the cases where r1≈ r2 and/ or r1r2 ≈ 1. In these cases small deviations in would more strongly affect the relative magnitudes of r1 and r2, which could have significant implications for engineering design purposes. For example, in the case where r1≈ r2, the errors introduced through first principles calculations could predict the existence of an azeotrope where one does not exist, and vice versa. We propose the investigation of these more difficult systems as future work.
In this work we have demonstrated that the transition states for surrogate polymer reactions can be obtained using computational chemistry. We have discovered that qualitative results are consistent with experiment, namely, that the addition of 1,3-butadiene is preferred in copolymerization with vinyl chloride. Furthermore, we have shown that the size of the basis set has only a slight effect on the activation energies, reactivity ratios, and polymer composition. In addition, we found that a smaller surrogate provides similar results at a fraction of the computational cost. Finally, the ab initio approach overestimated the relative reactivity of 1,3-butadiene, yielding a higher composition than experimentally observed.