Journal of Plant Biochemistry & Physiology
Open Access
ISSN: 2329-9029
ISSN: 2329-9029
Research Article - (2015) Volume 3, Issue 3
Floral organ development influences plant reproduction and crop yield. However, mechanisms underlying the development of floral organs in specific group of species like grasses remain unclear. To understand how palea was formed, we identified a retarded-palea2 (rep2) mutant, which showed that the palea was degenerate, and the lemma was crooked and just like sickle shaped. Genetic analysis confirmed that the rep2 mutant phenotype was due to a single recessive gene mutation. F2 population derived from the rep2 mutant crossed with Oryza sative subsp. Japonica Taigeng16, was used for molecular mapping of the REP2 gene. Using simple sequence repeats (SSR) and insertion-deletion (Indel) markers, the REP2 gene was fine mapped into a 12.9 kb physical distance on chromosome 9, where two open reading frames were predicted. Sequence analysis indicated that a 10-bp-deletion was found in LOC_Os09g24480 between 8PW33 and the rep2 mutant. The rice RETARDED PALEA1 (REP1) gene was in this locus. Thus, we suspected that a 10-bp deletion in the rep2 mutant caused a frame shift and premature translational termination, and led to the functional alteration of the REP2 gene.
Keywords: Rice (Oryza sative L. Subsp. indica); Rep2 mutant; Molecular marker; Gene mapping; Floral development
The flower is the reproductive organ of angiosperm plants, and its formation occurs through different steps. Firstly, the fate of the floral meristem is specified through the activity of floral meristem genes; secondly, the floral meristem is patterned into the whorls of organ primordia through the activity of floral organ identity genes; thirdly, the floral organ identity genes activate downstream effectors, which specify various tissues and cell types that constitute different floral-organ types [1]. Since some genes involved in flower development have been cloned from the model plants, rapid progress has been made in elucidating the molecular mechanisms regulating flowering [2,3]. Intensive molecular and genetic analyses in those species, notably Arabidopsis thaliana, and snapdragon (Antirrhinum majus), establish the ABC model [4,5]. According to this model, three classes of homeotic genes control the floral organ formation. A-class genes alone specify sepal formation, Aand B-class genes determine petal identity, B-class genes in combination with C-class genes together regulate stamen development, and C-class genes alone specify the innermost whorl, the carpel [6,7]. Although the floral model is proved by some studies in different plants [3,8-10] many questions remain unanswered. For example, the molecular mechanism underlying its floral organ development has not been fully investigated [11], how the target gene is identified by floral homeotic proteins, what is the target of floral homeotic proteins [12,13]. The grasses (Poaceae) is one of the largest monocot families with ~10 000 species, including many important cereal crops such as rice, maize, and barley [14,15]. Grass species have highly specialized flowers that differ from those of eudicots. A typical monocot, such as rice, a single spikelet consists of two pairs of sterile glumes (rudimentary glumes and empty glumes) and one floret, and a single floret is comprised of one lemma, one palea, two lodicules (the equivalent of petals), six stamens, and one pistil [16,17], but the surrounding structures, lodicules, lemma and palea, are unique to grasses [17,18]. In the eudicots, molecular genetic and morphological studies have revealed that lodicules and stamens are organs homologous to petals and stamens [2,19-21]. However, the origin and mechanism of lemma and palea development have long been controversial. Some researchers think of the lemma and palea as sepals, whereas others consider them as additional bracts because of their similar cellular patterns [19]. Hence, it is necessary to identify more mutants related to the development of lemmas and paleas and to isolate these relevant genes. However, to date, there are very few examples of molecular characterization of palea and lemma development in rice. In this study, we analyzed a rice mutant, retarded-palea2 (rep2), which had defects in several floral organ identities, including the palea identity, and in floral meristem determinacy. We also reported the investigation and comparison of the morphological features of the floral organs between the rep2 mutant and WT, genetic analysis of the mutant trait, and mapping of the gene for the rep2 locus to a small physical region. We isolated the rep2 gene by map-based cloning, and the rep2 gene encoded a TCP family transcription factor.
Plant materials
The Indica rice 8PW33 and Japonica Taigeng16 were kept in the Rice Research Institute, Fujian Academy of Agricultural Sciences, China. The retarded-palea2 mutant with the background of Indica cultivar 8PW33, was obtained through a screen of a M2 population treated by 60Co γ-ray, and was designated as rep2. About 300 plants in M1 population and 3000 plants in M2 population were grown at Fuzhou Experimental Station in Fujian Academy of Agricultural Sciences in April 2006 and in April 2007, respectively. The Indica cultivar, 8PW33, was derived from a progeny of a cross between Minghui 86 and Dongnanhui 307, which was designated as WT in this paper. In April 2011, the rep2 mutant was crossed with WT, 9311 and Taigeng16 at Sanya Experimental Station in Hainan Province. The F1 seeds, WT and the rep2 mutant were sown at Fuzhou Experimental Station in Fujian Province in June 2011 and all F2 seeds were harvested in October 2011. The F2 seeds, rep2 and 8PW33 were planted at Sanya Experimental Station in Hainan Province in April 2012. Plant height, panicle number per plant, spikelet number per panicle, flag leaf length, flag leaf width and setting percentage were measured at maturity in October 2011. The segregation ratios of mutants versus WT were investigated at flowering stage. All plants were grown according to standard commercial practices, with spacing of 13.3 cm between plants within each row and 26.4 cm between rows, and field management essentially followed normal agricultural practices.
Morphological and pollen fertility observation
In September 2011, 30 florets of mutant were selected randomly and the components of each floret were separated by forceps, and the types of florets were investigated. Images of florets were recorded by the Olympus digital anatomy microscope SZ61-SET. Estimation of pollen fertility was based on the I2-KI stain method [22]. Pollen was placed on slides with 1% I2-KI solution, and nipped into pieces using forceps to make the pollen grains spill out. The pollen fertility was examined under an optical microscope according to the morphology and staining gradation. The pistil fertility was investigated by the seed-setting rate of self-crossing and hybridization.
Construction of mapping population
The mapping population was constructed by crossing the rep2 mutant (Indica) with Taigeng16 (Japonica), and the F2 mapping population was generated from the self-cross of F1 population. Totally, 1808 mutant plants in F2 population were selected for fine mapping.
Microsatellite analysis
SSR (simple sequence repeats) primers were synthesized referring to the published rice database (http://www.Gramene.org/microsat/ssr. htm1). Indel (insertion-deletion) markers were designed according to the sequences comparison results of the genome sequences of Japonica (cv. Nipponpare) [23] with Indica (cv. 93-11) manually [23]. The BAC clone’s sequences of Japonica and Indica were aligned, and primers were designed using Primer premier 5.0 based on the polymorphism region between the two rice subspecies and the polymorphic markers were used for gene mapping.
PCR amplification and marker detection
Plant DNA was extracted from the frozen leaves of rice plants using the CTAB method [24] with minor modifications. For PCR amplification of markers, each 20 μL reaction mixture contained 50 ng DNA, 5 μmol of each primer, 10× PCR buffer (100 mM Tris (pH 8.3), 500 mM KCl, 15 mM MgCl2, 2 μg gelatin), 250 μM of each dNTP and 0.5 U of Taq polymerase. Amplification was performed with the following program: 5 min at 94°C, 35 cycles of 1 min at 94°C, 1 min at 55°C, and 2 min at 72°C, with a final extension of 5 min at 72°C. Amplified PCR products were resolved by electrophoresis in 3% agarose gels with ethidium bromide staining or 8% polyacrylamide denaturing gels with silver-staining for SSR markers [25].
Bulked segregant analysis
Bulked segregant analysis was used to search for markers linked to the target gene. A mutant DNA pool was constructed with the DNA extracted from leaves of 15 mutants randomly selected from the F2 population. SSR markers distributed in the rice genome were used to detect linkage, with DNAs extracted from the rep2 mutant and Taigeng16 used as a control. The band type of the markers linking with the mutant gene was the same as that of the rep2 mutant.
Molecular mapping of the rep2 gene
The band patterns of the mutant (rep2 rep2) and Taigeng16 (rep2 rep2) were recorded as 1 and 3, respectively, whereas 2 was used to denote the heterozygote (rep2 rep2). In this study, linkage analysis between the rep2 locus and the SSR markers was conducted using MAPMAKER version 3.0 software [26] and map distances were estimated with MapDraw V2 [27]. At the same time, the linkage map was basically the same as reported [28].
Bioinformatics analysis
Candidate genes were predicted according to the available sequence annotation databases (http://rice.plantbiology.msu.edu/; http://www. tigr.org/). DNA and amino acid sequences were used for a complete alignment using Clustal X version 1.81.
Main agronomic characteristics of rep2
To elucidate the genes that control the development of rice flowers, we screen for comparisons of phenotypes between the rep2 mutant and WT. The results showed that the rep2 mutant showed some special traits (Table 1). For example, the rep2 mutant is taller with wider flag leaf, higher with plant height and more spikelets than WT, which shows difference at 0.05 probable levels or significant difference at 0.01 probable levels. Especially, we observe that the seed setting rate of the rep2 mutant (48.4%) is lower than that of WT (92.6%) (Table 1), which shows significant difference at 0.01 probable levels.
Phenotype investigation of the rep2 mutant
WT and the rep2 mutant show indistinguishable phenotypes in their vegetative stage; however, their spikelets are different from booting stage to maturity (Figure 1a-1d). A wild type rice floret consists of a palea, a lemma, two lodicules, six stamens and a pistil with two stigmas, and these organs are all normal. In rep2 floret, the palea is degenerate, and the lemma is crooked, which is just like sickle shaped. Close examination of the base of the spikelet revealed that the degenerate palea in rep2 mutation was formed at the booting stage (Figure 1a). In addition, no abnormality was detected in other spikelet organs such as the pistil and stamens, suggesting that the rep2 mutation specifically affects the palea development. The rep2 mutant flowers were male fertile and female fertile. Microscopy analysis indicated that 95.75% of pollen grains of the mutated plants are normal (Figure 1g). When artificially pollinated with the WT pollens, about 300 germinative seeds were yielded from 400 flowers of 3 panicles on one plant, suggesting that the rep2 mutant was basically female fertile.

Figure 1: Phenotypes of WT (image on the right of each panel) and the rep2 mutant (left)
Genetic analysis of the gene for the rep2 trait
To determine whether rep2 was controlled by a single gene or multiple genes, the rep2 mutant was crossed with 9311and WT. All F1 hybrids showed normal phenotypes, and all F2 populations showed normal Mendel’s segregation (Table 2). Segregation of WT and the mutant type plants fitted a 3:1 segregation ratio in the two F2 populations (χ2=0.330~0.688, P>0.05) (Table 2). So these results indicated that the mutant phenotype was controlled by a single recessive gene.
| Crosses | F1 phenotype | F2 population | χ2(3:1) | P | ||
|---|---|---|---|---|---|---|
| Normal plants | Palea degradation | Total plants | ||||
| rep2/WT | Normal type | 156 | 46 | 202 | 0.330* | 0.5-0.75 |
| rep2/9311 | Normal type | 135 | 55 | 190 | 0.688* | 0.25-0.5 |
Table 2: Segregations of F2 populations crossed by the rep2 mutant.
Preliminary molecular mapping of the rep2 gene
At first, the polymorphisms between the rep2 mutant and Taigeng16 were examined with 324 pairs of SSR primers from the RM series, of which 206 pairs exhibited polymorphism. Using BSA (bulked segregant analysis) method analysis, these 206 primer-pairs, the rep2 mutant, Taigeng16, and 15 mutant plants from the F2 population were then used for linkage analysis between markers and the mutant gene. The result suggested that SSR marker RM3796, RM6051, RM6854and RM566 were linked to rep2 locus, and the polymorphic linkage-markers RM3796, RM6051, RM6854and RM566 were used to survey 189 F2 plants derived from rep2 mutant × Taigeng16. Based on the segregation data and linkage map analyzed with MAPMAKER version 3.0 and Map Draw V2.1 software, the rep2 locus was located between markers RM6051and RM6854 on chromosome 9, at a respective distance of 3.6 cM and1.2 cM (Figure 2a), Then, ten pairs of new primers in the proximal region of the rep2 locus were used for mapping. Among them, four primers, RM24282, RM24301, RM24323, and RM24334 showed polymorphism between the rep2 mutant and Taigeng16 (Table 3). Then the rep2 gene was preliminarily mapped between molecular markers RM24301 and RM24323 in the terminal region of chromosome 9, at a respective distance of 0.7 cM and 0.4 cM (Figure 2b).

Figure 2: Genetic and physical maps of the REP2 gene.
| Markername | Markertype | Sequence of forward primer | Sequence of reverse primer | Locations |
|---|---|---|---|---|
| RM5657 | SSR | 5’-TATGTGCATTTGTAAGGTGA-3’ | 5’-GCTTTAGATTATTGAGCGAG-3’ | OJ1261_A08 |
| RM24240 | SSR | 5’-ATGCAACCTCCTCCATCATAAGG-3’ | 5’-TGCTGCTCCTACCTCACTCACC-3’ | B1040D06 |
| RM24260 | SSR | 5’-ACTAAAGGTCCCTAGATGA-3’ | 5’-TAAAGATGTTGGCTATGTC-3’ | P0435D08 |
| RM24275 | SSR | 5’-TATAGCAAGAGCCATAGC-3’ | 5’-CTACCAACCCAGATGAAC-3’ | P0650H04 |
| RM24282 | SSR | 5’-TTGTGGTTATTTGGCTGTC-3’ | 5’-CGAACTGTTAAACGATGTG-3’ | OSJNBb0014M19 |
| RM24301 | SSR | 5’-GAGCTGGATGTCCTCGAACG-3’ | 5’-GACCACCTCTCCAAGCTCACC-3’ | OSJNBa0084L05 |
| Indel-9-1 | Indel | 5’-TATGTGCATTTGTAAGGTGA-3’ | 5’-GCTTTAGATTATTGAGCGAG-3’ | OJ1261_A08 |
| Indel-9-3 | Indel | 5’-TCTTGCATTGACACCTCTTGAGC-3’ | 5’-AGTCCCAACAACTGGAAGAGAGG-3’ | P0465E03 |
| Indel-9-4 | Indel | 5’-TGACGTGTCTAGGTCCATAATGC-3’ | 5’-TTTCCTGTTCCGTTTGTCAGG-3’ | OJ1585_D02 |
| Indel-9-17 | Indel | 5’-GCCACGGCCAGTTCAACTCC-3’ | 5’-CCGTCCGGATCTTGCTGTGC-3’ | OJ1294_G06 |
| Indel-9-8 | Indel | 5’-GAACAGAGGAGGAGATCGAGAGG-3’ | 5’-CTTCTTGGGAGATGCAGAAATGG-3’ | OJ1294_G06 |
| Indel-9-19 | Indel | 5’-ATCACCCGCCCATTATGCTACCC-3’ | 5’-GATGTGGTCACCCTGACATGTGG-3’ | OJ1294_G06 |
| Indel-9-23 | Indel | 5’-GAACACGAGCGTCTTCTTCACC-3’ | 5’-GTTGGCTTTGATCGATGTGTCG-3’ | OJ1294_G06 |
| Indel-9-12 | Indel | 5’-CGATGTGTCGTCGTCGTC-3’ | 5’-AGCTCCTCGTGCAGAAGAAG-3’ | OJ1294_G06 |
| Indel-9-13 | Indel | 5’-ACCCAACTACGATCAGCTCG-3’ | 5’-CTCCAGGAACACGCTCTTTC-3’ | P0643D11 |
| Indel-9-16 | Indel | 5’-TCCACTTCATCTTCTCAACC-3’ | 5’-CGGAGTAGATCAGTAGGATCG-3’ | P0643D11 |
| RM24323 | SSR | 5’-GTATATATCCGTGCGAATCACTCTCC-3’ | 5’-AACACAGCTCACGCCAGTTCC-3’ | P0707C02 |
| RM24334 | SSR | 5’-GAACGGTTTGAGGAAGAAGAAGACG-3’ | 5’-ATCCATCCACGACACACCATCC-3’ | P0668D04 |
Table 3: In Del and SSR molecular marker used for fine mapping of the REP2 gene.
Fine mapping of the rep2 gene
To map the gene to a smaller region, 1808 mutant individuals were identified from the F2 population derived from rep2 × Taigeng16. A higher precision map was constructed using published markers in the region between RM24301 and RM24323 (Figure 2c, Table 3). Seven polymorphic InDels were selected from 16 new InDels (Table 3). The Indel markers were designed from the publicly available rice genome sequences, and the likelihood of detecting polymorphism between the rep2 mutant and Taigeng16 was predicted by comparing sequences from Nipponbare (http://rgp.dna.affrc.go.jp/) and the Indica cultivar 93-11 (http://rice.genomics.org.cn/). All recombinants were genotyped using seven polymorphic markers within the above interval. Recombinant screening with nine markers (RM24301, Indel-9-1, Indel-9-3, Indel-9-4, Indel-9-8, Indel-9-12, Indel-9-13, Indel-9-16 and RM24323), which were more internal to the rep2 locus. The rep2 gene was precisely defined in an 80.0 kb region by Indel-9-4 and Indel-9-12 (Figure 2c). To delimit the gene to a smaller region, new polymorphic molecular markers were developed, and three polymorphic InDels were selected from 8 new InDels (Table 3). Recombinant screening with six other markers (Indel-9-4, Indel-9-17, Indel-9-8, Indel-9-19, Indel-9-21 and Indel-9-12), which were more internal to the rep2 locus, detected two, one, zero, two, two and six recombinants, respectively (Figure 2d). Thus, the rep2 gene was precisely defined in a 12.9 kb region by Indel-9-17 and Indel-9-19.
Candidate genes in the 12.9 kb region
There are two annotated genes (LOC_Os09g24460 and LOC_ Os09g24480) in the 12.9 kb region, according to the available sequence annotation databases (http://ricegaas.dna. affrc.go.jp/; http://www.tigr. org/). The two annotated genes have a corresponding full-length cDNA. LOC_Os09g24460 is an expressed protein, and LOC_Os09g24480 encodes a TCP family transcription factor.
Sequence analyses of the rep2 gene
To investigate which gene was responsible for the mutation phenotype, sequencing of two genes in WT and the rep2 mutant revealed that a 10-bp-deletion was found in LOC_Os09g24480 between WT and the rep2 mutant (Figure 3), while no difference in the LOC_Os09g24460 between WT and the rep2 mutant was observed. Thus, we concluded that the LOC_Os09g24480 locus corresponded to rep2. Interestingly, the RETARDED PALEA1 (rep1) gene, encoding TCP gene family members in defining the diversification of floral morphology [33], was in this locus. The phenotype characters of the rep2 were very similar to that of rep1 (Figure 1a-1e). According to the phenotypic resemblance, mapping and sequencing analysis, we suspected that rep2 was probably allelic to rep1. The analysis of the ORF region showed that the rep2 gene (LOC_Os09g24480) had corresponding full length cDNAs of 777bp (Figure 4). In rep2, a 10- bp deletion was found in LOC_Os09g24480, causing a frame shift and premature translational termination (Figure 4). Thus, mutation as such would be expected to significantly alter the functions of the protein.

Figure 3: Sequence comparison between WT and REP2 in LOC_Os09g24480.
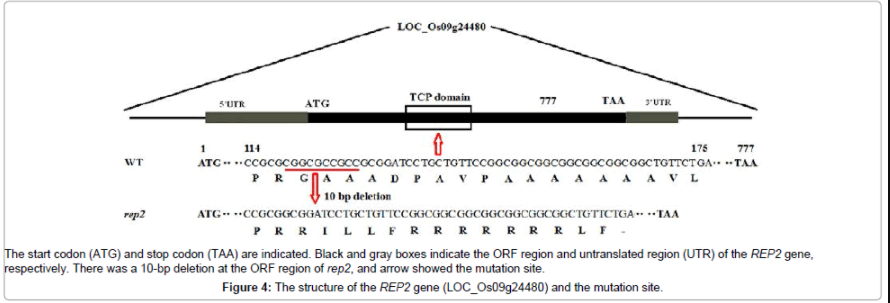
Figure 4: The structure of the REP2 gene (LOC_Os09g24480) and the mutation site.
Homology analyses of rep2 genes from other species
To gain insight into the function of rep2, we generated homology analyses between rice and Arabidopsis (Figure 5) according to available sequence annotation databases (http://rice.plantbiology.msu.edu/cgibin/ gbrowse/rice/). In Arabidopsis, an ortholog of rep2, AT3G18550, which belonged to the TCP family, was identified to encode TCP family transcription factor. As shown in Figure 5, rep2 and AT3G18550 had high homology. Interestingly, a 10-bp deletion was found in the rep2 mutant (Figure 4, 5), which caused a frame shift and premature translational termination, and destroyed the conservative region, leading to a functional alteration of the rep2 gene. Thus, we suspected that the phenotype of the rep2 mutant would be caused by the functional alteration of the TCP structural protein.
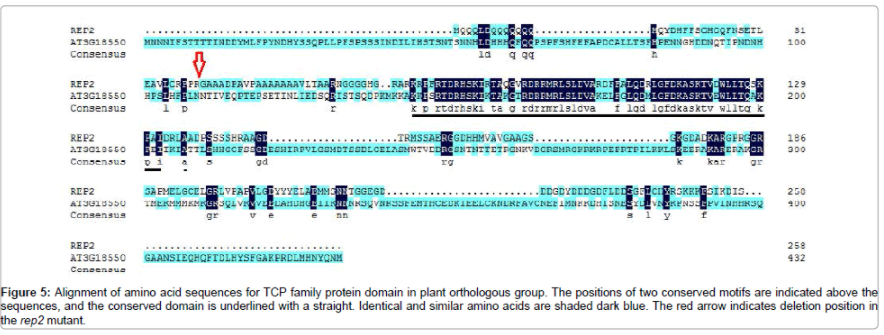
Figure 5: Alignment of amino acid sequences for TCP family protein domain in plant orthologous group. The positions of two conserved motifs are indicated above the sequences, and the conserved domain is underlined with a straight. Identical and similar amino acids are shaded dark blue. The red arrow indicates deletion position in the rep2 mutant.
The rep2 mutant exhibits unique qualities
Facilitated by the recent developments in genome sequencing, molecular markers and bioinformatics, an impressive number of the floral organ identity genes are fine mapped and cloned in the past 20 years, and it is more and more important to identify mutants related to the development of lemmas and paleas in cloning the floral organ genes. In rice, several lemma and/or palea defective mutants have been reported; for example, leafy lemma and calcaroides in barley [29], lhs1 [30], dh1 [31], sl1 [32], rep1 [33], mof1 [34], dep [35], pal1 [36], tob1 [37], bls1[2] and slp1 [38] in rice. In the present study, we have characterized and identified the rep2 mutant. The rep2 gene was located on chromosome 9, and sequence analysis showed that a 10-bp deletion was found in LOC_Os09g24480 (the rep1 locus) among rep2 and WT. Meanwhile, the rep2 mutant and the rep1 mutant possessed many similar characters. These results indicated that the rep2 was allelic to rep1. The rep2 is not only an epigenetic allele described in rice but also exhibits unique qualities compared with the rep1. For example, the rep2 mutant shows different traits compared with WT (Table 1). Interestingly, we observed that the seed setting rate of the rep2 mutant (48.4%) was lower than that of WT (92.6%) (Table 1). The previous observations showed that the rep2 mutant had normal stamen and pistil, and why the rep2 mutant had lower seed setting rate? We further observation showed that the rep2 mutant had at least 3 stamens exposed outside during flowering and pollination (Figure 2c). Therefore, we speculate that stamens of the rep2 mutant exposed outside, which are difficult to reach on the pistil stigma, and lead to the rate of decline.
| Main traits | WT | The rep2 mutant | Difference |
|---|---|---|---|
| Plant height (cm) | 99.3 | 119.6 | -20.3** |
| Productive panicles per plant | 6.8 | 7.3 | -0.5 |
| Flag leaf length (cm) | 55.0 | 58.4 | -3.4 |
| Flag leaf width (cm) | 2.00 | 2.80 | -0.8* |
| Spikelets per panicle | 152.3 | 255.3 | -102.0** |
| Seed setting rate (%) | 92.6 | 48.4 | 44.2** |
Table 1: Comparison of main agronomic traits between the rep2 mutant and WT.
rep2 is a TCP family member
In this article, using a map-based cloning strategy we isolated the rep2 gene, which encoded a putative protein and belonged to a plantspecific TCP transcription factor family. These TCP family members have previously only been identified in angiosperms and have been shown to be essential in specifying plant morphology [39]. For example, TB1 in maize [40], OsTB1 [41], rep1[40] in rice, and AtTCP12/ BRC1 proteins in Arabidopsis [42] are associated with controlling zygomorphic floral development. However, a key question is whether there are TCP genes that control the diversification of floral asymmetry in grasses. Through genetic and molecular studies, some researchers have addressed this point in rice [33]. Therefore, this finding theref ore extended the function of the TCP gene family members in defining the diversification of floral morphology in grasses, and suggested that a common conserved mechanism controlling floral zygomorphy by CYClike genes existed in both eudicots and monocots [33]. In summary, the rep2, which is allelic to rep1, plays an important role in establishing palea identity and controlling the diversification of floral asymmetry in rice. Therefore, further molecular study on the rep2-like proteins in the relative grass family will help us to facilitate elucidate whether the conserved pathway exists in controlling other grass floral zygomorphy.
This work was supported in part by the National Natural Science Foundation of China (No. 31201197), Technology Innovation Team of Fujian Academy of Agricultural Sciences (No. CXTD-1-1301), Special Fund for Agro-scientific Research in the Public Interest of Fujian Province (No. 2014R1021-4), Fujian Provincial Natural Science Foundation of China (No. 2015J01108), and Technology Great Item of Fujian Province (2013NZ0002-2).