Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Review Article - (2014) Volume 2, Issue 5
Aspirin responsive erythromelalgia is the presenting symptom of thrombocythemia in patients with essential thrombocythemia (ET) and polycythemia vera (PV). Skin punch biopsies taken from the affected areas of erythromelalgia and acrocyanotic compications show typical arteriolar inflammation, fibromuscular intimal proliferation and thrombotic occlusions. If left untreated both microvascular and major thrombosis frequently do occur in thrombocythemia in ET and PV patients, but can easily be cured and prevented by low dose aspirin. The stratification as low, intermediate and high thrombotic risk in the retrospective Bergamo studies has been performed in ET patients not treated with aspirin. The risk of thrombosis in aspirin treated ET and PV is not age dependent when on low dose aspirin, and does recur when not on low dose aspirin during follow-up. The persistence of the Bergamo definition of low, intermediate and high thrombotic risk in the 2012 International Prognostic Score of Thrombosis in ET (IPSET) is applicable for JAK2 mutated ET and PV patients not on aspirin and has led to significant overtreatment with hydroxyurea. MPN disease burden in patients with JAK2V617F positive ET and PV is related to JAK2 allele burden and associated leukocytosis, thrombocytosis, constitutional symptoms and splenomegaly. Low dose peglyated interferon (PegasysR, 45 μg/ ml once per week or every two weeks) is becoming the first line myeloreductive treatment option to postpone the useof hydroxyurea as long as possible. Von Willebrand factor (VWF) mediated platelet thrombi formation, as well as increased proteolysis of the VWF multimers in one and the same patient do occur simultaneously or in sequence leading to the paradoxical occurrence of thrombosis at platelet count aboe 400x109/L and bleeding at platelet counts above 1000x109/L due to an acquired von Willebrand disease type 2A.
Keywords: Erythromelalgia; Hemorrhages; Arterial thrombosis; Thrombocythemia; Polycythemia vera; Essential thrombocythemia; Hemorrhagic thrombocythemia; Thrombotic thrombocythemia; Aspirin; Myeloproliferative disorders
Mitchel first reported a syndrome of redness and burning pain in the extremities, which were labeled erythromelalgia [1]. The relief of pain for several days after a single dose of aspirin (500 mg) is specific and can be used as a diagnostic criterion for erythromelalgia [2]. We discovered that aspirin responsive erythromelalgia is causally related to thrombocythemia in patients with essential thrombocythemia (ET) and polycythemia vera (PV) at platelet counts around and above 400x10/L [3,4]. By demonstratin that erythromelalgia in thrombocythemia disappeared for 2 to 4 days after a single dose of aspirin (500 mg). This could be attributed to the irreversible inactivation of platelet cycooxygenase activity for 2 to 4 days by acetyl-salicylic acid (aspirin). As sodium salicylic acide has no cyclo-oxygenase inhibiting acticity on platelets and has no effect at all on erythromelalgia we concluded in 1985 that aspirin responsive erythromelalgia in thrombocythemia is caused by platelet mediated microvascular infammatrory and thrombosis in ET and PV patients [3-6].
The present study reviews the original description of the broad spectrum of microvascular, thrombotic and hemorrhagic manifestations in ET and PV patients related to peripheral blood cell counts, bone marrow features, and burden of myelopoliferative neoplasm (MNP) disease.The pathophysiology of platelet-mediated arterial thrombosis and bleeding in ET and PV patients is discussed and interpreted in view of the recent literature and updated treatment recommendations for the prevention of platelet-mediated thrombosis and bleeding in thrombocythemia and by reduction of MPN disease burden by pegylated interferon or hydroxyurea.
Prospective Clinical Studies on Erythromelalgia and Acrocyanotic Toes or Fingers in ET and PV
The broad spectrum of clinical features of erythromelalgia and peripheral, cerebral and coronary ischemic events in 24 patients with myeloproliferative thrombocythemia (11 ET and 13 PVT patients were collected by the Rotterdam MPN Study Group in the period between 1975 and 1981 (Table 1) [3,4]. Complaints of erythromelalgia consisted of painful red to cyanotic toes or fingers. The pain may vary from prickling, pins and needle stick sensation to a severe burning pain. The involved areas of fore foot toe or fingers are always swollen and painful on palpation and touch (Figures 1 and 2) [3-6]. At time of burning pain severe attacks of tingling prickling may occur [3,4]. The burning pain of erythromelalgia increased on walking compelling the patient to rest with the involved extremity elevated. After rest with the involved extremity elevated, the erythromelalgic redness may disappear but the extremity remains swollen, acrocyanotic and painful on touch. Fingertips or toes remain red, swollen and severely painful like a burn. Acrocyanotic spots in erythromelalgic areas were present in 8 cases indicating localized ischemia (Table 1) [3-6]. In one ET patient (A11), warm painful swollen redness of the big toe was accompanied by a cold dark blue cyanotic non-swollen third toe. In a third patient (A24, PV) who presented with typical pronounced erythromelalgia of the fingertipss was followed by cold, swollen acrocyanosis of 4 fingers of the left hand (aspirin responsive blue finger and black toe syndrome in PVT, Figure 1) [1-4]. In ET erythromelalgia already develops at pflaftefleft counft fin excess off 350 fto 400x10 /L. WThen sympftomaftfic at ‘normal’ platelet count around 350x109/L in PV in remission by phlebotomy alone platelet count usually rise to values above 400x109/L during aspirin treatment [3,4]. The time lapse between first complaints of typical erythromelalgia and the diagnosis of ET or PV elapsed from a few months to 5 years, (mean 2,25) years [3,4]. One ET patients (A2) had a very long history of erythromelalgia complicated by peripheral gangrene, atypical TIAs and recurrent coronary ischemic events. Seven cases with longstanding erythromelalgic signs and symptoms developed ischemic complications: acrocyanosis with blister formation of the big toe (A1), a necrotic spot in the tip of a toe in 4 (A1, A1, A4, A20) [3,4]. In PV case 20 with red congestion of the big and second toe and a necrotic spot of the third toe bone marrow biopsy from the anterior iliac crest was complicated by wound dehiscent and subcutaneous ecchymosis. Sympathectomy because of ischemic complication was not effective in 5 cases with blue black toes and digital gangrene (thromboangiitis obliterans) in the absence of infection and inflammation in 3 (A4, A5, A10), and necrosis of the distal phalanx of a toe in 2 (A2, A9). Angiogram in one ET case (A1) with erythromelalgia complicated by digital gangrene showed abrupt occlusions of toe arteries without any evidence of vascular pathology of the supplying arteries [5,6]. The sequential occurrence of aspirin responsive erythromelalgic, ocular and cerebral microvascular disturbances in relation to platelet counts and the effects of platelet reduction during 15 years follow-up has been describe in detail 6. Transient cerebral ischemic events in one PV patient (A24) was followed by overt erythromelalgia in the finger tips, which were left untreated and complicated by acrocyanosis of 4 fingers of the left hand showing abrupt arterial occlusions of the hand arcade and of supplying digital finger arteries of the 4 involved acrocynotic swollen fingers (Figure 1) [5,6]. Strikingly, the arterial pulses of arteriae dorsales pedis and arteriae tibiales remain normal in all cases of erythromelalgia complicated by peripheral acrocyanosis or digital gangrene. Complete relief of erythromelagic and/or ischemic pain and restoration of the ischemic acral circulation disturbances (thrombo-angiitis obliterans) in ET and PVT patients could be obtained by long-term treatment with low-dose aspirin (250 mg/day). In the five patients with erythromelalgia complicated by digital grangrene (Table 1) [3,4] experienced the favorable effect of aspirin, but it took about one or two weeks before all ischemic symptoms completely disappeared [5,6]. If left untreated, cold cyanotic fingertip or toe tips may develop, remain painful and resistant to aspirin very likely due to irreversible digital arteriolar occlusion. Standard therapy with oral anticoagulants (warfarin) and reduction of hematocrit to normal by bloodletting in PVT did not affect the erythromelalgic ischemic symptoms and acrocyanotic digital ischemia and gangrene (thrombo-angiitis obliterans).
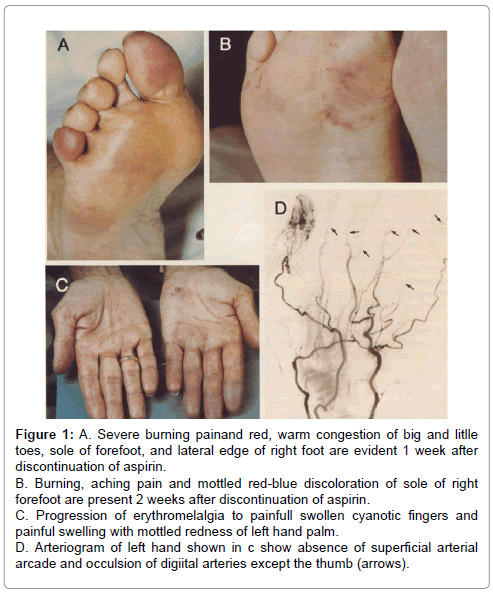
Figure 1: A. Severe burning painand red, warm congestion of big and litlle toes, sole of forefoot, and lateral edge of right foot are evident 1 week after discontinuation of aspirin. B. Burning, aching pain and mottled red-blue discoloration of sole of right forefoot are present 2 weeks after discontinuation of aspirin. C. Progression of erythromelalgia to painfull swollen cyanotic fingers and painful swelling with mottled redness of left hand palm. D. Arteriogram of left hand shown in c show absence of superficial arterial arcade and occulsion of digiital arteries except the thumb (arrows).
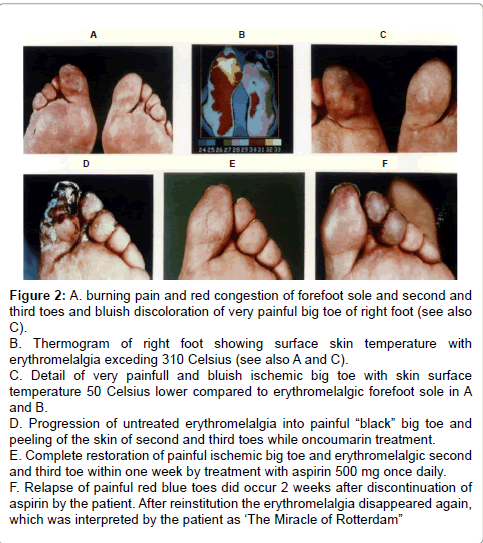
Figure 2: A. burning pain and red congestion of forefoot sole and second and third toes and bluish discoloration of very painful big toe of right foot (see also C). B. Thermogram of right foot showing surface skin temperature with erythromelalgia exceding 310 Celsius (see also A and C). C. Detail of very painfull and bluish ischemic big toe with skin surface temperature 50 Celsius lower compared to erythromelalgic forefoot sole in A and B. D. Progression of untreated erythromelalgia into painful “black” big toe and peeling of the skin of second and third toes while oncoumarin treatment. E. Complete restoration of painful ischemic big toe and erythromelalgic second and third toe within one week by treatment with aspirin 500 mg once daily. F. Relapse of painful red blue toes did occur 2 weeks after discontinuation of aspirin by the patient. After reinstitution the erythromelalgia disappeared again, which was interpreted by the patient as ‘The Miracle of Rotterdam”
| Number | Gender | Platelets/ | Hb / Ht | Erythromelalgia: | TIA/ACS | History ETT | |||
| Diagnosis | Age / yrs | 109/L | mmol/L % | Toes | Fingers | Skin | Gangrene | months | |
| A1 ET | F 62 | 792 | 10.3/50 | bilateral | present | 45 | |||
| A2 ET | M 39 | 887 | 10.0/51 | bilateral | present | TIA/ACS | 154 | ||
| A3 ET | F 42 | 911 | 8.9/47 | unilateral | yes | ACS | 60 | ||
| A4 ET | M 47 | 614 | 8.0/39 | unilateral | present | TIA | 12 | ||
| A5 ET | M 46 | 939 | 8.3/40 | unilateral | present | 4 | |||
| A6 ET | M 33 | 742 | 9.8/45 | TIA/ACS | 20 | ||||
| A7 ET | M 45 | 567 | 9.5/46 | bilateral | yes | ACS | 60 | ||
| A8 ET | M 39 | 875 | 8.8/43 | bilateral | bilateral | yes | 30 | ||
| A9 ET | M 51 | 690 | 8.8/45 | bilateral | present | 20 | |||
| A10 ET | M 40 | 1440 | 8.6/43 | unilateral | present | 30 | |||
| A11 ET | F 72 | 1435 | 9.4/46 | unilateral | 30 | ||||
| A12 PVT | M 63 | 1932 | 11.1/56 | bilateral | 24 | ||||
| A13 PVT | F 75 | 1800 | 12.1/62 | unilateral | yes | 3 | |||
| A14 PVT | M 61 | 952 | 8.3/45 | bilateral | 0 | ||||
| A15 PVT | M 53 | 636 | 7.7/39 | unilateral | present | 36 | |||
| A16 PVT | F 60 | 1065 | 13.4/68 | bilateral | 48 | ||||
| A17 PVT | F 49 | 728 | 10.9/57 | bilateral | 1 | ||||
| A18 PVT | M 66 | 1035 | 12.2/64 | bilateral | 18 | ||||
| A19 PVT | M 71 | 1320 | 13.3/70 | unilateral | 2 | ||||
| A20 PVT | M 65 | 1300 | 11.9/65 | unilateral | present | 24 | |||
| A21 PVT | F 55 | 1085 | 12.2/61 | unilateral | 4 | ||||
| A22 PVT | F 59 | 708 | 11.0/59 | bilateral | yes | 3 | |||
| A23 PVT | F 74 | 959 | 13.1/72 | unilateral | unilateral | 24 | |||
| A24 PVT | M 71 | 609 | 12.5/66 | unilateral | 6 | ||||
Table 1: Erythromelalgia and microvascular disturbances in 24 patients with ETT (11 ET and 13 PV) [3].
In the 1970s very little was known about the histopathology of erythromelalgia in thrombocythemia as the opportunity to examine tissue from the involved extremities of typical fresh erythromelalgia and of its acrocyanotic complications was not or only scarcely available [7]. We performed between 1975 and 1980 skin punch biopsies from erythromelalgic areas and from acrocyanotic areas in thrombocythemia patients (ET and PV) [7]. The histological appearances of a skin punch biopsy from anarea of recently relapsed erythromelalgia 1 week after discontinuation of aspirin, show arteriolar leasions without involvement of venules, capillaries of nerves (Figure 3) [7]. Arterioles showed pronounced proliferation and degeneration in their vessel walls. The lining endothelial cells are swollen some with large nuclei. The inner layer of the arteriolar vessel wall was thickened by intimal proliferation of cells with narrowing of the lumen. With factor VIII antisera an affected arteriole with intimal thickening shows an inner layer of fluorescencent endothelial cells (Figure 4) and with the antiserum against smooth muscle cells strong flurescence was seen in the proliferated subendothelial intimal cell zone (fibromuscular intimal proliferation, Figure 3) [7]. Slight intravascular and perivascular infiltration with inflammatory cells and some perivascular fibrosis is present. The internal elastic lamina is seen to be splitted by proliferating smooth muscle cells (fibromuscular intimal proliferation, Figure 3) [7].
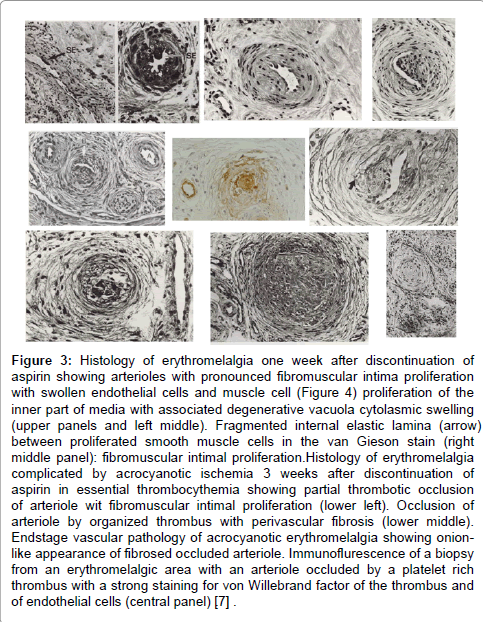
Figure 3: Histology of erythromelalgia one week after discontinuation of aspirin showing arterioles with pronounced fibromuscular intima proliferation with swollen endothelial cells and muscle cell (Figure 4) proliferation of the inner part of media with associated degenerative vacuola cytolasmic swelling (upper panels and left middle). Fragmented internal elastic lamina (arrow) between proliferated smooth muscle cells in the van Gieson stain (right middle panel): fibromuscular intimal proliferation.Histology of erythromelalgia complicated by acrocyanotic ischemia 3 weeks after discontinuation of aspirin in essential thrombocythemia showing partial thrombotic occlusion of arteriole wit fibromuscular intimal proliferation (lower left). Occlusion of arteriole by organized thrombus with perivascular fibrosis (lower middle). Endstage vascular pathology of acrocyanotic erythromelalgia showing onionlike appearance of fibrosed occluded arteriole. Immunoflurescence of a biopsy from an erythromelalgic area with an arteriole occluded by a platelet rich thrombus with a strong staining for von Willebrand factor of the thrombus and of endothelial cells (central panel) [.7] .
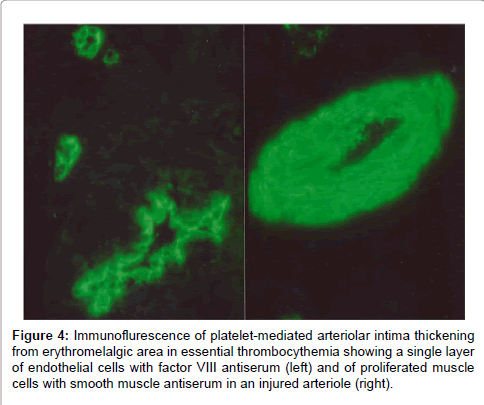
Figure 4: Immunoflurescence of platelet-mediated arteriolar intima thickening from erythromelalgic area in essential thrombocythemia showing a single layer of endothelial cells with factor VIII antiserum (left) and of proliferated muscle cells with smooth muscle antiserum in an injured arteriole (right).
The histopathological lesions from relapsed erythromelalgia complicated by bluish spots 3 weeks after discontinuation of aspirin, show arteriolar lesions of fibromuscalr intimal proliferation occluded by an organized fresh von willebrand factor rich platelet thrombi (Figure 3). The skin wedge excision from an acrocynotic erythromelalgic toe mainly shows arterioles with fibromuscular intimal proliferation with narrowing of the lumen and on top of that occluded arterioles by fresh organized thrombus is seen in erythromelalgic areas complicated by acrocyanosis (Figures 3 and 5). The thickened arteriolar microvasculature ultimately show occluded arterioles by thrombi with perivascular fibrosis without inflammatory cell infiltration give rise to onion-like structues, in which the internal lamina elastic appears to be absent and the external elastic membrane is fragmented (Figure 3) [7].
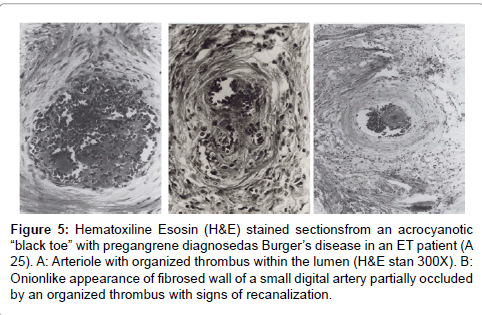
Figure 5: Hematoxiline Esosin (H&E) stained sectionsfrom an acrocyanotic “black toe” with pregangrene diagnosedas Burger’s disease in an ET patient (A 25). A: Arteriole with organized thrombus within the lumen (H&E stan 300X). B: Onionlike appearance of fibrosed wall of a small digital artery partially occluded by an organized thrombus with signs of recanalization.
Erythromelalgic Cerebral and Ocular Ischemic Manifestations in ET and PV
In the original cohort of 24 patients with myeloproliferative thrombocythemia (11 ET, 13 PVT) aspirin responsive migraine-like atypical transient ischemic attacks (MIA) and acute coronary syndrome (ACS) were observed in eight patients (5 ET, 3 PVT, Table 1) [3,4]. Recurrent sudden attacks of unconsciousness lasting for a few minutes did occur in 3 patients with erythromelalgia (A2, A3, A16). Migraine like headache followed by recurrent transient hemiparesis lasting for half an hour (migraine accompagne) was associated with disorientation in time [8] in one patient with a history of myocardial infarction. One case suffered from severe migraine–like headache followed by nausea and emesis after discontinuation of aspirin (A7). On case with a history of myocardial infarction suffered from recurrent transient ischemic attacks (A6). In 1993 we reported on the presenting transient neurologic and ocular manifestation in ET patients [9] and subsequently reviewed the literature [10,11]. The transient neurologic and ocular symptoms in ET are featured by migraine like atypical transient cerebral or ocular ischemic attacks (MIAs) [9-11]. The main neurologic symptoms in ET are poorly localized cerebral symptoms of unsteadines, unstable gait, or attacks of sudden falling without loss of consciousness or weakness, focal transient cerebral symptoms of mono- or hemiparesis and focal transient visual symptoms of monocular blindness [9]. The cerebral or visual symptoms are accompanied by migraine-like dull or throbbing unilateral or bilateral headaches, usually lasting for several hours. None of the patients had a history of migraine. Global visual symptoms include scintillating scotomas and blurred vision. All transient neurlogic and visual attacks had a sudden onset and occurred gradually rather than all at once and lasted for a few seconds to several minutes. The progression had both a rapid (within seconds) spreading of visual symptoms and a sequential occurrence of one symptom after another; for instance migraine-like visual symptoms followed by dysarthria, followed by weakness [9]. Progressfion off ftransfienft Themfiparesfis fto sftroke or permanent blindness did occur if not treated with aspirin. Routine screening with brain CT, carotid duplex, ultrasonography, or cardiac investigations did not reveal any obvious coexistent abnormality of cause of MIAs in affected ET and PV patients [9-11].
Three cases (1ET and 2 PV) developed necrotic toes and were diagnosed as thrombo-angiitis obliterans (Buerger’s disease) of the toes, which were already amputated by a surgeon before referral to us because of thrombocythemia for expert evaluation and treatment recommendation.
The 1980 Rotterdam Clinical and Pathological Criteria for ET and PV
Focusing between 1975 and 1980 on the causal relation between erythromelalgia and thrombocythemia in ET and PV patients, we were able to document the very early stage of ET by the use of the Rotterdam Clinical and Pathological (RCP) criteria for ET and PV (Table 2) [3,4]. Since 1975 we found platelets in excess of 400 x 109/L, and an increase of clustered enlarged megakaryocytes in a bone marrow biopsy material diagnostic for ET (Table 2) [3,4]. Iron stain of bone marrow histology specimens from ET and PV patients were performed with Prussian blue reagents. The RCP criteria of ET and PV were determined in 1980 based on careful prospective documentation of peripheral blood and bone marrow histology from bone marrow biopsy material. Platelets in excess of 400×109/L, and an increase of clustered enlarged megakaryocytes in a bone marrow biopsy material was found to be diagnostic for ET and excluded reactive thrombocytosis (Table 2). The minimum criterion for the diagnosis according to the PVSG in 1975 was 1000x109/L [12], which was reduced to 600x109/L in 1986 [13,14].
| S. No | Criteria |
| A | Major criteria |
| A1Persistent platelet count in excess of 400x109/L. | |
| A2 Increase and clustering of large megakaryocytes in bone marrow biopsy | |
| B | Confirmative criteria |
| B1 Presence of large platelets in a peripheral blood smear | |
| B2 Absence of any underlying disease for reactive thrombocytosis and normal ESR. | |
| B3No or slight splenomegaly on palpation or scan ( | |
| B4 Increase of LAP-score and no signs of fever or inflammation | |
| C | Exclusion criterion |
| Ph+ chromosome and any other cytogenetic abnormality in blood or bone marrow cells | |
| B. The 1980 RCP major (A) and minor (B) criteria for prefibrotic PV3 | |
| A1 Raised red cell mass. Male >36 ml/kg, female >32 ml/kg: PVSG 197514 | |
| A2 Increased erythrocyte count above 6x1012/L. | |
| A3 Increase in bone marrow biopsy ofclustered, large pleomorphic megakaryocytes with hyperlobulated nuclei and increased cellularity due to increased megakaryopoiesis erythropoiesis or typically trilinear mega-erythro-granulopoiesis. typical PV bone marrow excludes erythrocytosis618,19. | |
| B1 Thrombocythemia, persistant increase of platelet >400x109/L | |
| B2 Leukocytosis, leucocyte count >109/L and low erythrocyte sedimentation rate | |
| B3Raised leukocyte alkaline phosphatase (LAP) score >100, absence of infection | |
| B4 Splenomegaly on palpation or on isotope/ultrasound scanning | |
| A1or A2 plus A3 and none of B establishes erythrocythemic PV | |
| A1 or A2 plus A3 plus one of B establishes PV and excludes erythrocytosis |
Table 2: The Rotterdam Clinical and pathological (1980 RCP) criteria of essential thrombocythemia (ET) defined and used between 1975-1980 [3].
Between 1975 and 1980 we used bone marrow histology as the pathognomonic clue to PV on top of the 1975 PVSG criteria for the diagnosis of polycythemia vera [14,15]. Erythrocyte volume (red cell mass RCM) was measured using Cr51 (natriumchromate) labeled autologous erythrocyte1. The RCP modifications in 1980 of the 1975 PVSG criteria for PV14 include 4 main changes (Table 2) [3,4]. First, the major criterion O2-saturation of >92% is deleted since a bone biopsy differentiates between PV and erythrocytosis9. Second; splenomegaly is replaced by bone marrow histology as a major criterion (A3, Table 2). Third, the RCP diagnostic set used splenomegaly as a minor criterion (Table 3a). Fourth, we skipped raised B12 (>900 ng/L) or raised B12 binding capacity (>2200 ng/L) as completely irrelevant for the diagnosis of early and overt stage PV (Table 2).
| Number | Diagnosis | Gender | Spleen | Spleen | Platelets | Hb | Ht | Erythroc | BM iron |
| Age yrs | scan cm | 109/L | mmol/l | 1012/L | stain | ||||
| A1 | ET | F 60 | np | 10.6 | 792 | 10.4 | 0.49 | 6.7 | neg |
| A2 | ET | M 39 | np | 9.8 | 887 | 10 | 0.51 | 6 | pos |
| A3 | ET | F 42 | . | 5 | 911 | 8.9 | 0.47 | 5.4 | pos |
| A4 | ET | M 47 | p | 13.5 | 614 | 8 | 0.39 | 4.5 | - |
| A5 | ET | M 46 | np | 10.5 | 939 | 8.3 | 0.4 | 4.4 | pos |
| A6 | ET | M33 | p | 14.3 | 742 | 9.8 | 0.49 | 5.8 | pos |
| A7 | ET | M 45 | np | 11.2 | 567 | 9.5 | 0.46 | 5.2 | pos |
| A8 | ET | M39 | np | 10.8 | 875 | 8.8 | 0.43 | 4.9 | pos |
| A9 | ET | M51 | np | 11.2 | 690 | 8.8 | 0.45 | 5.5 | pos |
| A10 | ET | M40 | p | 14.4 | 1440 | 8.6 | 0.43 | 4.7 | pos |
| A11 | ET | F 72 | p | 14.8 | 1435 | 9.4 | 0.46 | 6.1 | neg |
| A12 | PTV | M 63 | np | 8 | 1932 | 11.1 | 0.56 | 6.5 | neg |
| A13 | PTV | F 75 | np | . | 1800 | 12.1 | 0.62 | 7.6 | neg |
| A14 | PTVb | M 61 | . | 5 | 952 | 8.3 | 0.45 | 5.6 | neg |
| A15 | PTVb | M 53 | p | 13.1 | 636 | 7.7 | 0.39 | 5.4 | neg |
| A16 | PTV | F 60 | p | 14.5 | 1065 | 13.4 | 0.68 | 7.9 | neg |
| A17 | PTV | F 49 | p | 15.8 | 728 | 10.9 | 0.57 | 7.5 | neg |
| A18 | PTV | M 66 | p | 15.9 | 1035 | 12.2 | 0.64 | 7.1 | neg |
| A19 | PTV | M 71 | p | 15.3 | 1320 | 13.3 | 0.7 | 6.4 | neg |
| A20 | PTV | M 65 | p | 14.9 | 1300 | 11.9 | 0.65 | 7.6 | neg |
| A21 | PTV | F 55 | p | 14.1 | 1085 | 12.1 | 0.61 | 7.1 | neg |
| A22 | PTV | F 59 | p | 12.5 | 708 | 11 | 0.59 | 7.5 | neg |
| A23 | PTV | F 74 | p | 14.4 | 959 | 13.1 | 0.72 | 9.1 | neg |
| A24 | PTV | M 71 | p | 16.6 | 609 | 12.5 | 0.66 | 9.9 | neg |
| B1 | PV/HT | M 50 | p | 2316 | 10.2 | 0.52 | 6.3 | neg | |
| B2 | PV/HT | F 86 | p | 19 | 2975 | 5.3 | 0.32 | 4.4 | neg |
| B3 | ET/HT | M 72 | np | 11.4 | 1503 | 6.3 | 0.31 | 3.5 | pos |
| B4 | ET/HT | F 45 | np | 13.8 | 1074 | 8.1 | 0.41 | 4.6 | neg |
| B5 | ET/HT | M 51 | np | 10.1 | 1080 | 8.3 | 0.42 | 4.9 | - |
| B6 | PV/HT | M 61 | np | 11.4 | 737 | 9.9 | 0.49 | 5.1 | neg |
Table 3a: Clinical and peripheral blood data in 14 ET and 16 PTV patients (Source Michiels Thesis 1981[:1 ]; A 1 / 2 4 complicated by erythromelalgia and B1/6 hemorhagic thrombocythemia (HT) [3]. ET: Essential Thrombocythemia, PTV: Polycythemia Thrombocythemia Vera. b= Treated With Busulphan, np: Not Palpable, p=Palpable Source . Michiels J.J. Thesis, Erythromelalgia and Thrombocythemia, Rotterdam 198:160-163.
Prospective Laboratory Investigations and Bone Marrow Features of Thrombocythemia in ET and PV
Platelet-mediated arteriolar inflammation and platelet thrombi (erythromelalgia) did not occur in reactive thrombocytosis, which strongly indicates not only a quantitative but also a qualitative disorder of platelet hyperfunction in thrombocythemia [3,4,8,10]. This unknown platelet defect in thrombocythemia in ET and PV paients is postulated by Michiels and co-workers to lead to an increased risk of microvascular thrombotic complication and microvascular and macrovascular thrombotic complication in thrombocythemia of various MPNs [16- 20]. Phlebotomy alone in PV significantly reduced the incidence of macrovascular complication at normal hematocrits of 0.40 or less. A hematocrit of 16-20].
In the period of January 1975 to December 31, 1980, we prospectively studied the laboratory and bone marrow features in 30 consecutive early prefibrotic stage MPN patients, who presented with erythromelalgic thrombotic thrombocythemia (ETT), 14 ET and 16 PV patients (Table 3b) [3,4]. The clinical features of erythromelalgia caused by platelet-mediated arteriolar inflammation and thrombosis in thrombocythemia of the ET and PV cases have been reported in great detail in the Annals of Internal Medicine, 19854. The mean age of 30 ETT patients (ET and PV) was 56.7 (range 33-96) years. Eleven of 14 ET patients had platelet counts below 1000x109/L, in which the RCP diagnosis of ET (Table 2) would have been overlooked by the PVSG criteria at that time. Spleen size on scan was slightly increased in 5 of 14 ET and in 13 of 16 PV patients. Leukocyte count counts was increased (>10x109/L) in 5 out of 14 patients with ET and in 14 of 16 PV patients. LAP score was increased (>100) in 12 out of 14 ET and in all PV patients.
| Number | Leukoc | LAF | RCM Ery | ESR | BM | BM1 | BM1 | BM M/E | BM iron |
| 109/L | score | volume | megakar | cellularity | reticuline | ratio | stain | ||
| A1 ET | 10 | 183 | 31 | 2 | 1+ | N | N | 2.1 | neg |
| A2 ET | 9 | 155 | 0 | 1+ | N | N | 2.9 | pos | |
| A3 ET | 8 | 109 | 1 | 2+ | 2+ | 1+ | 3.3 | pos | |
| A4 ET | 9 | 101 | 4 | ||||||
| A5 ET | 16 | 128 | 26 | 2 | 1+ | N | N | 3 | pos |
| A6 ET | 7 | 139 | 6 | 1+ | N | N | 3.1 | pos | |
| A7 ET | 8 | 127 | 3 | 1+ | N | N | 2.9 | pos | |
| A8 ET | 10 | 38 | 3 | 2+ | 1+ | 1+ | pos | ||
| A9 ET | 10 | 103 | 52 | 1+ | 1+ | 1+ | 3.2 | pos | |
| A10 ET | 11 | 60 | 5 | 1+ | N | N | 4 | pos | |
| A11 ET | 13 | 113 | 32 | 3 | 2+ | 1+ | 2+ | 1.9 | neg |
| A12 PV | 10 | 207 | 59 | 75 | 1+ | 2+ | 1+ | 1.1 | neg |
| A13 PV | 28 | 193 | 2 | 2+ | 2+ | N | 1.7 | neg | |
| A14 PV | 13 | 236 | 4 | 2+ | 2+ | 2+ | 1.1 | neg | |
| A15 PV | 11 | 103 | 63 | 2+ | 1+ | 1+ | 1.6 | neg | |
| A16 PV | 17 | 243 | 45 | 3 | 2+ | 2+ | 1+ | 1.2 | neg |
| A17 PV | 8 | 186 | 60 | 1 | 1+ | 1+ | 1+ | 0.6 | neg |
| A18 PV | 14 | 184 | 63 | 2 | 2+ | 1+ | 1+ | 2.2 | neg |
| A19 PV | 16 | 219 | 50 | 1 | 2+ | 2+ | 1+ | 3.3 | neg |
| A20 PV | 18 | 128 | 38 | 3 | 2+ | 1+ | 1+ | 1.2 | neg |
| A21 PV | 13 | 170 | 43 | 1 | 2+ | 2+ | 2+ | 2.1 | neg |
| A22 PV | 17 | 168 | 42 | 0 | 2+ | 2+ | 2+ | 1.3 | neg |
| A23 PV | 9 | 219 | 54 | 2 | 2+ | 2+ | 1+ | 0.5 | neg |
| A24 PV | 18 | 215 | 82 | 0 | 2+ | 2+ | 1+ | 1.3 | neg |
| B1 HT | 46 | 0 | 2+ | 2+ | 2+ | neg | |||
| B2 HT | 24 | 235 | 38 | 1 | 2+ | 2+ | 2+ | 0.6 | neg |
| B3 HT | 8 | 49 | 4 | 2+ | N | 1+ | 2.2 | pos | |
| B4 HT | 7 | 105 | 7 | 1+ | N | N | 2.5 | neg | |
| B5 HT | 10 | 47 | 2 | 2+ | 2+ | 2+ | - | ||
| B6 HT | 8 | 121 | 36 | 1+ | N | 1+ | 1.8 | neg |
Table 3b: Peripheral blood and bone marrow data in 14 ET and 16 PV patients (Source Michiels Thesis 1981 [1]): A1/24complicated by erythromelalgia, and B1/6 hemorrhagic thrombocythemia (HT) [3]. Source. Michiels J.J. Thesis, Erythromelalgia and Thrombocythemia, Rotterdam 1981:160-163.
Increase of clustered large megakaryocytes and a normoblastic erythropoiesis (decreased M/E ratio) was present in bone marrow smears and biopsies of 14 ET and 16 PV patients (Table 3b, Figures 6 and 7). The myeloid/erythrocyte (M/E) ratio was shifted towards increased erythropoiesis in half of ET and most PV patients. The mean M/E ratio in 14 patients with early stage ET was 1.95 (range 0.6-4.0) as compared to 1.53 (range 0.6-3.3) in 16 PV patients. A normocellular bone marrow picture with increase of clustered pleomorphic megakaryocytes and no increase of cellularity (Figure 5) was seen in 7 of 14 ET and in one of 16 PV patients. A moderate increase of cellularity (1+=60-80%) in the bone marrow due to increased erythropoiesis (=decreased M/E ratio) leading to a ET/PV picture was seen in 3 ET and 4 PV patients (Figure 7). A typical PV bone marrow picture with pronounced increase of cellularity (2+=80-100%) due to predominantly increased erythropoiesis or trilinear hypercellular hematopoiesis (megakaryo/ erythro/granulocytic: panmyelosis Figure 6) was seen in 2 of 14 ET patients and in 11 of 16 PV. These results indicate that bone marrow histopathology on its own is not reliable to clearly differentiate between ET and PV but appeared to be a powerful tool to differentiate ET and PV from all variants of primary or secondary erythrocytosis and reactive thrombocytosis with a sensitivity and specificity near to 100% [3,4]. The morphology of large pleomorphic megakarocytes were not different in ET and PV except that pleomorphism of megakaryocytes was more pronounced in the hypercellular (80-100%) bone marrows of PV and ET patients with advanced myeloproliferative disease (Figure 7) [3,4]. We could conclude in 19813 that RCP defined ET and PV patients show overlapping bone marrow pictures with increase of pleomorphic clustered megakaryocytes and varying degrees of increased erythropoisesis at time of diagnosis (Figures 6 and 7).
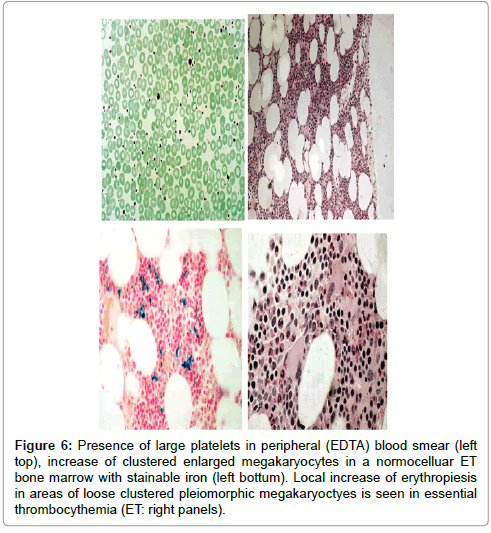
Figure 6: Presence of large platelets in peripheral (EDTA) blood smear (left top), increase of clustered enlarged megakaryocytes in a normocelluar ET bone marrow with stainable iron (left bottum). Local increase of erythropiesis in areas of loose clustered pleiomorphic megakaryoctyes is seen in essential thrombocythemia (ET: right panels).
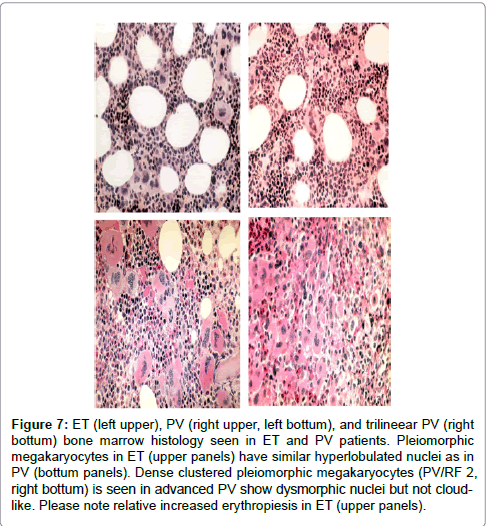
Figure 7: ET (left upper), PV (right upper, left bottum), and trilineear PV (right bottum) bone marrow histology seen in ET and PV patients. Pleiomorphic megakaryocytes in ET (upper panels) have similar hyperlobulated nuclei as in PV (bottum panels). Dense clustered pleiomorphic megakaryocytes (PV/RF 2, right bottum) is seen in advanced PV show dysmorphic nuclei but not cloudlike. Please note relative increased erythropiesis in ET (upper panels).
Erythromelalgic Thrombotic Thrombocythemia (EET) and Hemorrhagic Thrombocythemia (HT)
In the period we reported the erythromelalgic, thrombotic and hemorhagic manifestations in 50 consecutive cases of thrombocythemia in 30 ET and 20 PV patients 10: 16 ET and 15 PV patients suffered from erythromelagic thrombotic thrombocythemia (ETT). Besides erythromelalgia 16 thrombocythemia patients suffered from additional transient arterial ischemic or thrombotic complications involving thebrain or heart : transient cerebral neurologic ischemic attacks (TIA) in 3, and migraine-like atypical TIAs in 8 ; myocardial infarction in 2; amourosis fugax in 2 and venous thrombosis was not observed. Hemorrhagic thrombocythemia (HT) was diagnosed in 8 patients without erythromelalgia. The presenting features of HT were bruises and ecchymoses in 4, recurrent epistaxis in 2 bleeding after tooth extraction in 2 and gatsrointestinal bleeding in 2. Paradoxical occorrence of erythromelalgia and bleeding symptoms (ETTT plus HT) was observed in 5 thrombocythemia patients. The laboratory data at time of presentation in 30 ET and 20 PV patients are shown in Figure 8. Hemoglobin levels were normal in ET and elevated in PV. Leukocyte counts were normal or increased in ET and PV. The leukocyte alkaline phosphatase score was much more elevated in PV than in ET (Figure 8).
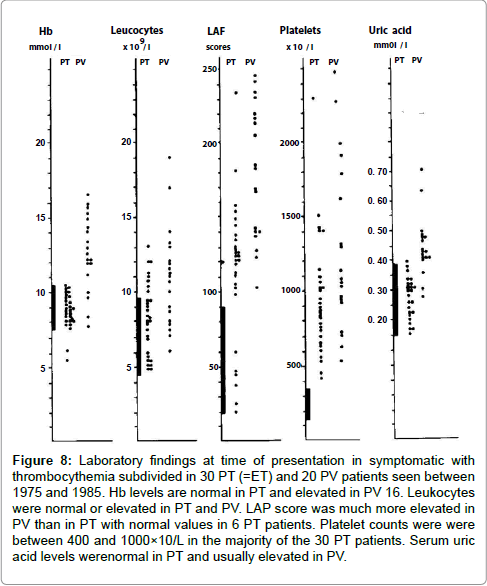
Figure 8: Laboratory findings at time of presentation in symptomatic with thrombocythemia subdivided in 30 PT (=ET) and 20 PV patients seen between 1975 and 1985. Hb levels are normal in PT and elevated in PV 16. Leukocytes were normal or elevated in PT and PV. LAP score was much more elevated in PV than in PT with normal values in 6 PT patients. Platelet counts were were between 400 and 1000×10/L in the majority of the 30 PT patients. Serum uric acid levels werenormal in PT and usually elevated in PV.
Seventeen of 31 patients with erythromelalgic thrombotic thrombocythemia had platelet counts below 1000x109/L with the lowest count of 420x109/l (Figure 8) [10]. Platelet counts in 8 HT patients and 5 ETT/HT patients with paradoxal erythromelalgia and bleeding had platelet counts between 850 to 3000x109/L and were in excess of 1000x109/L in 12 of 13 thrombocythemia patients (Figure 9). Budde et al discovered that increased platelet count far above 1000x109/L in HT was associated with an acquired von Willebrand syndrome type 2A [21,22]. Michiels and co-workers prospectively documented in ET and PV patients the details of paradoxical occurrences of erythromelalgic microvascular thrombotic manifestation and spontaneous mucocutaneous bleeding at platelet count above 1000x109/L during long-term follow-up caused by an acquired von Willebrand syndrome type 2A, which disappeared after correction of platelet count to normal (Figures 10-12) [20,23-27].
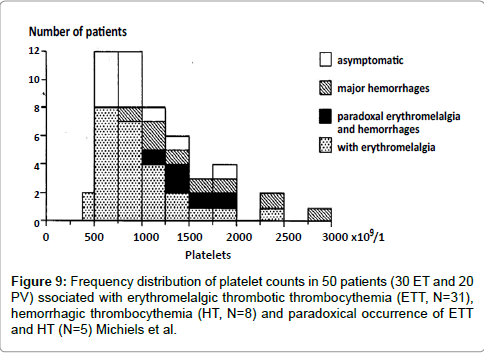
Figure 9: Frequency distribution of platelet counts in 50 patients (30 ET and 20 PV) ssociated with erythromelalgic thrombotic thrombocythemia (ETT, N=31), hemorrhagic thrombocythemia (HT, N=8) and paradoxical occurrence of ETT and HT (N=5) Michiels et al.
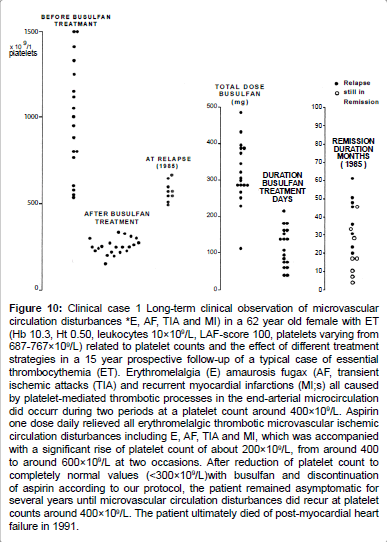
Figure 10:Clinical case 1 Long-term clinical observation of microvascular circulation disturbances *E, AF, TIA and MI) in a 62 year old female with ET (Hb 10.3, Ht 0.50, leukocytes 10×109/L, LAF-score 100, platelets varying from 687-767×109/L) related to platelet counts and the effect of different treatment strategies in a 15 year prospective follow-up of a typical case of essential thrombocythemia (ET). Erythromelalgia (E) amaurosis fugax (AF, transient ischemic attacks (TIA) and recurrent myocardial infarctions (MI;s) all caused by platelet-mediated thrombotic processes in the end-arterial microcirculation did occurr during two periods at a platelet count around 400×109/L. Aspirin one dose daily relieved all erythromelalgic thrombotic microvascular ischemic circulation disturbances including E, AF, TIA and MI, which was accompanied with a significant rise of platelet count of about 200×109/L, from around 400 to around 600×109/L at two occasions. After reduction of platelet count to completely normal values (< 300×109/L)with busulfan and discontinuation of aspirin according to our protocol, the patient remained asymptomatic for several years until microvascular circulation disturbances did recur at platelet counts around 400×109/L. The patient ultimately died of post-myocardial heart failure in 1991.
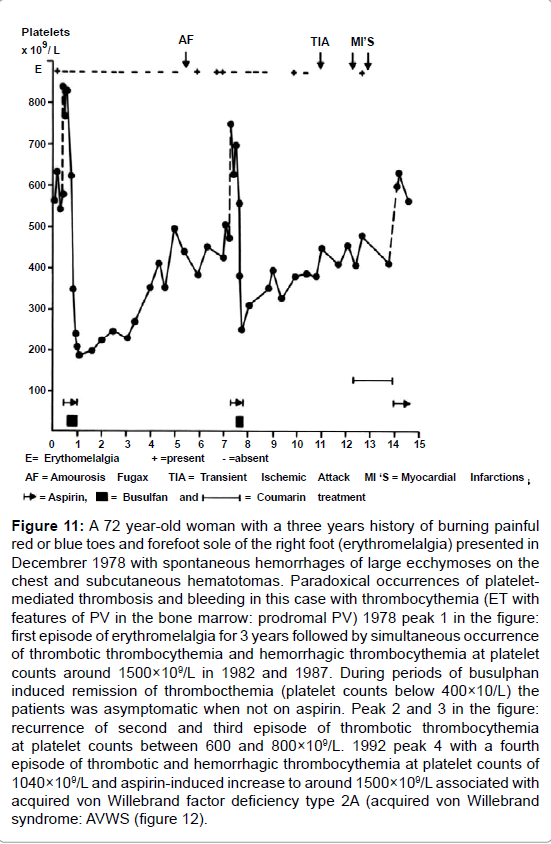
Figure 11: A 72 year-old woman with a three years history of burning painful red or blue toes and forefoot sole of the right foot (erythromelalgia) presented in Decembrer 1978 with spontaneous hemorrhages of large ecchymoses on the chest and subcutaneous hematotomas. Paradoxical occurrences of plateletmediated thrombosis and bleeding in this case with thrombocythemia (ET with features of PV in the bone marrow: prodromal PV) 1978 peak 1 in the figure: first episode of erythromelalgia for 3 years followed by simultaneous occurrence of thrombotic thrombocythemia and hemorrhagic thrombocythemia at platelet counts around 1500×109/L in 1982 and 1987. During periods of busulphan induced remission of thrombocthemia (platelet counts below 400×10/L) the patients was asymptomatic when not on aspirin. Peak 2 and 3 in the figure: recurrence of second and third episode of thrombotic thrombocythemia at platelet counts between 600 and 800×109/L. 1992 peak 4 with a fourth episode of thrombotic and hemorrhagic thrombocythemia at platelet counts of 1040×109/L and aspirin-induced increase to around 1500×109/L associated with acquired von Willebrand factor deficiency type 2A (acquired von Willebrand syndrome: AVWS (figure 12).
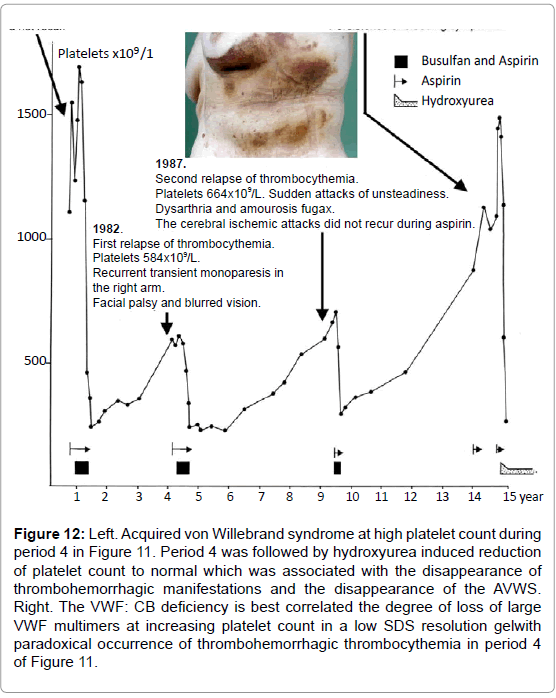
Figure 12: Left. Acquired von Willebrand syndrome at high platelet count during period 4 in Figure 11. Period 4 was followed by hydroxyurea induced reduction of platelet count to normal which was associated with the disappearance of thrombohemorrhagic manifestations and the disappearance of the AVWS. Right. The VWF: CB deficiency is best correlated the degree of loss of large VWF multimers at increasing platelet count in a low SDS resolution gelwith paradoxical occurrence of thrombohemorrhagic thrombocythemia in period 4 of Figure 11.
Platelet Lowering Agents in ET: Low Dose Busulphan to Control Platelet Number and ETT Symptoms
The European Organization on Research and Treatment of Cancer (EORTC) conducted phase III study in previously untreated PV patients *n=293). Previously untreated PV patients were randomized for P32 and busulfan in the years 1967-1978 [28]. The hematologic objective response criteria were given in terms of hematocrit values between 0.42 and 0.47. With a median follow-up of 9 years, busulfan treatment (mean dose 310 mg) was better than P32 (mean dose 5.9 mCi) [28]. There were fewer thrombotic episodes with busulfan than with P32 due to better control of the thrombocythemia with busulfan: 25 of 149 treated with P32 died of vascular complication compared to 8 of 145 treated with busulfan, but the incidence of acute leukemia after a median follow-up of 9 years was equally low and not statistically different between the two agents (2/140 P32 versus 3/145 busulphan [28]. Overall survival at 10 years was significantly better with busulphan (74%) than with P32 (55%). This difference in overall survival was due to a much higher frequency of vascular accidents in the P32 arm because of a poorer control of hemoglobin values and platelet counts. Consequently, twenty five of 149 patients treated by P32 died from vascular complications not on aspirin compared to 8 of 145 PV patients treated by busulphan not on aspirin [28].
The London Myeloproliferative Study Group of Wetherley-Meinet al used the alkylating agent buslphan more judiciously as an effective and safe platelet reductive agent for the control of thrombocythemia in PV [29], and for the control of platelet cout in symptomatic cases of ET30. In the London ET study, Wetherley-Mein and co-workers reported on the effective treatment with busulphan in 37 cases (mean age 60.5, range 30-89 years) with PVSG defined ET with platelet counts between 615-2450x109/L [30]. The vascular occlusive symptoms at presentation in 19 patients were painful feet and toes (erythromelalgia) in 20, foot/leg ulcers in 4, atypical transient neurologic or ocular ischemic manifestations in 11, major thrombosis in 8. Hemorrhagic thrombocythemia (HT) had occured in 8 at platelet counts in excess of 1000x109/L. Reduction of platelet count by low dose busulphan to less than 400x109/L and no aspirin resolved all vascular occlusive disease and the disappearance of bleeding [30]. Cox regression analysis revealed two prognistically important features in ET: age had a strong inverse correlation with survival and vascular occlusive symptoms correlated with a better survival. The number of death was 2.1 times than of a comparable control group and death from thrombosis was not significantly different from the number expected [30]. The natural history was featured by progression of ET into PV occurred in 9% and to myelofibrosis in 24% with death from myelofibrosis markedly increased [30].
From 1974 to 1986 the Rotterdam MPN Working Group treated 20 symptomatic ET patients with aspirin and one course of busulaphan The symptomatic microvascular thrombotic circulation disturbances ranged from erythromelalgia (N=18) , atypical and typical transient ischemic attacks (N=6), and acute coronary syndromes (N=3) were [31,32]. Mean platelet count before busulphan treatment was 1009x109/L,( range 545 to >1525, Figure 13). One course of busulfan treatment was started at a dose of 4 to 6 mg/day at platelet counts >1000x109/L (Figure 10). At platelet count just below 1000x109/L busulfan dose was reduced to 4 or 2 mg/day under carful blood cell counts every week. Busulphan was discontinued in all 20 ET cases when platelet counts reached the upper limit of normal (350x10/L (Figure 13). At time of complete remission of ET, aspirin was discontinued to test the hypothesis whether control number to normal was associated with cure and no recurrence of erythromelalgic circulation disturbances. As shown in Figure 9 the mean platelet count before and after busulfan treatment was 1009x10/9L (range 545-1525) and 241 (range 159-315) x109/L respectively. At time of normal platelet count (<350x109/L) erythromelalgia did not recur after discontinuation of aspirin. The total dose burden of only one course of busulfan treatment ranged from 106-484 (mean 315) mg. The durarion of the busulfan course ranged from 44 to 218 (mean 116) days. All 20 patients with busulphan induced remission of ET remained asymptomatic after discontinuation of both busulfan and aspirin as long as platelet counts were normal for a follow-up period from 10 to 60 months (Figure 10). At time of relapse of ET (platelets >400x109/L) in 12; 8 became symptomatic (mainly erythromelalgia and atypical TIA in a few) at slight increased platelet count of 410, 450, 490, 500, 515, 545, 548 and 635x109/L indicating the need of low dose aspirin (100 mg), and 4 remained asymptomatic at platelet counts of 577, 600, 648 and 725x109/L (Figure 10). Based on these prospective intervention studies, we decided in 1986 to treat our ET patients with aspirin at time of presentation, during followup, and at relapse as long as platelet counts were between 400 and 1000+200x109/L. In 1986, hydroxyurea at that time became the drug of choice for clear indications to reduce platelet count from above to below 1000x109/L with continuation of low dose aspirin (Tables 4 and 5). At that time bleeding symptoms, aspirin side effects and platelet counts in excess of 1000 to 1500x109/L were clear indications for correction of platelet counts in ET and PV patients [10,31,32].
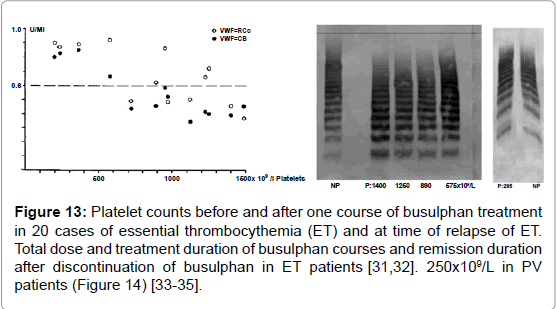
Figure 13: Platelet counts before and after one course of busulphan treatment in 20 cases of essential thrombocythemia (ET) and at time of relapse of ET. Total dose and treatment duration of busulphan courses and remission duration after discontinuation of busulphan in ET patients [31,32]. 250x109/L in PV patients (Figure 14) [33-35].
| Thrombotic complications | Bleeding complications | ||||
| Treatment Strategy | Person/yrs | Events (n) | person-years | Events (n) | person-yrs |
| Asymptomatic 14 Patients: | |||||
| Watchful waiting | 127 | 27* | 33.3 | 2 | 1.6 |
| Symptomatic 54 patients: | |||||
| Low-dose aspirin | 139 | 5 | 3.6 | 10*** | 7.2 |
| Platelet reduction | 113 | 10** | 8.9 | 2 | 1.8 |
| Low-dose aspirin + | 40 | 0 | - | 4 | 10 |
| platelet reduction | |||||
| Total | 419 | 42 | 18 | ||
Table 4: Incidence of thrombotic and bleeding complications in the prospective 1975-1996 Rotterdam study of 68 ET patients during a median follow-up of 6.7 years according to treatment strategy (Van Genderen et al 1997) [36,37]. Mean platelet count *=610, range 410-831×109/l at time of thrombotic event. **=624 ± 255×109/l at time of thrombotic event. ***=1737, range 661-3460×109/l at time of bleeding event (Table 5).
| Platelets: | Platelets: | Platelets: | Platelets: |
| 400-1500x 109/L | 400-1000x109/L | 400-1000x109/L | >1500x109/L |
| Low risk ETT/HT | Standard risk ETT | High risk ETT/HT | High ETT/HT risk |
| JAK2 wild type | JAK2V617F mutated | JAK2V617F mutated | All variants ET |
| Asymptomatic Microvascular dsturbances |
Microvascula r disturbances only* |
Major thrombosis Spontaneous bleeding or elicited by aspirin | >1000 x 109/L and minor thrombosis/ bleeding = high |
| No vascular risk | No vascular risk | Vascular risk(s) | No vascular risk |
| No bleeding risk | No bleeding risk | Bleeding risk | |
| All ages* | All ages* | Age >65 years* | All ages |
| Aspirin uncertain Wait and see? | Low dose aspirin 50 to 100 mg/day Intermediate risk | Platelet reduction to normal or near normal |
Platelet reduction to 9/l |
| Aspirin primary prevention? ET patients and their physician usually prefer the use of low dose aspirin | Microvascular disturbances and platelet count between 1,000 and 1,500 x109/l with clear indication aspirin*, ➜side effects platelet reduction | Continue aspirin* | When platelets <1,000 x 109/l continue or add aspirin |
Table 5: Low, intermediate and high thrombohemorrhagic risk stratification of ET, ET with features of early PV in blood and bone marrow (prodromal PV) and classical PV patients: a flexible approach towards therapeutic implications with reference to platelet counts [11]. *At platelet counts in excess of 1,000+250x109/l aspirin will usually elicit bleeding symptoms, which disappear after reduction of platelet counts to below 1,000 x 109/l with continuation of aspirin (Figure 4). If side effects of aspirin (gastritis, GIbleeding occur) reduce platelet to normal and consider indomethacin 25 mg TID.
The Spectrum of ETT and HT in ET and PV patients: Therapeutic Implications
In 1980 [33-35] Michiels reviewed 100 case histories of primary hemorrhagic thrombocythemia (HT) and 99 cases of erythromelalgic thrombotic thrombocythemia (ETT, 67ET and 32 PV). The bleeding manifestations in 100 HT cases ranged from gastrointestinal chronic occult blood loss, melena and hematemesis to mucocutaneous bruises, hematomas, ecchymoses, gum bleedings and secondary bleeding [33-35]. HT was usually associated with significant leukocytosis and splenomegaly and the platelet count at time of bleeding in 100 HT cases ranged from 800 to above 4000x109/L. The manifestations in 99 ETT (67 ET and 32 PV) patients included erythromelalgia, acrocyanosis, digital gangrene, amaurosis fugax, transient ischemic attacks, stroke, angina pectoris and myocardial infarction, superficial thrombophlebitis and deep vein thrombosis. The platelet count at time of ETT in ET and PV patients ranged from 400 to 2000x109/L in ET patients and from 350 to 1
ETT=Erythromelalgic Thrombotic Thrombocythemia. HT=Hemorrhagic Thrombocythemia; ET: Essential Thrombocythemia; PV=Polycythemia Vera.
VWF:RCo=Von Willebrand Ristocetine Cofactor Activity; VWF: CB=Von Willebrand Factor Collagen Binding.
The final follow-up of the Rotterdam MPD Working Group (1975-1995) on the efficacy and safety for the prevention and treatment of thrombotic complications in 68 ET patients seen in the Academic Hospital Rotterdam has been critically evaluated by Perry van Genderen et al (1997) [36,37] independent from the principal investigator (Michiels). In this prospective observational Rotterdam ET study the 68 consecutive ET patients were followed between 1975 to 1995 for a total of 419 patient years (Table 4) [36,37]. At time of presentation 54 patients had ET related microvascular thrombotic complications at platelet counts between 575 and 1031, mean 750x109/L and treated with low-dose aspirin, a platelet lowering agent (busulphan or hydroxyurea) or both. The 14 ET asymptomatic ET patients suffered from 27 thrombotic and two bleeding episodes occurred during a follow-up of 127 patient years; platelet counts at time of thrombotic event were between 410 and 831, mean 610x109/L. Only four of the initially 14 asymptomatic ET patients remained asymptomatic after at least 30 person-years follow-up. In the treatment group of low-dose aspirin alone, 5 thrombotic events during a follow-up of 179 patient years, and 10 bleeding episodes occurred in ET patients receiving low dose aspirin monotherapy during a follow-up of 139 patients years at platelet counts between 661 and 3460 (mean 1737)x109/l (Table 4). The main conclusion from this observation is that aspirin alone in ET at platelet counts above 1000x109/l in general daily practices is not safe enough in terms of bleedings elicited by aspirin indicating the need to add one of the non-leukemogenic platelet lowering agents pegylated interferon or anagrelide [36,37]. In patients receiving platelet lowering agents either busulfan or hydroxyurea and no aspirin, 10 thrombotic complications occurred during follow-up of 40 patient years at platelet counts above 400x109/L (624 ± 25x109/L, Table 4), indicating that inadequate control of platelet number in thrombocythemia of ET and PV patients is associated with a persistent high incidence of microvascular disturbances including MIAs when not on aspirin at plateletcounts above 350 to 400x109/L (Figure 14). In this landmark study of Van Genderen et al, age above 65 years was not a risk factor for thrombotic recurrences in ET when on low dose aspirin [36,37]. In the 1986 London study of 37 ET cases treated with low dose busulphan for the treatment of ETT and HT not on aspirin (mean age 60.5, range 30- 89 years ,platelet between 615-2450x109/L, Figure 14) [25] all vascular occlusive disease and bleeding resolved at time of platelet correction to normal [30]. Age had a strong inverse correlation with survival but vascular occlusive symptoms correlated with a better survival; progression of ET into PV occurred in 9% and to myelofibrosis in 24% with death from myelofibrosis markedly increased [30].
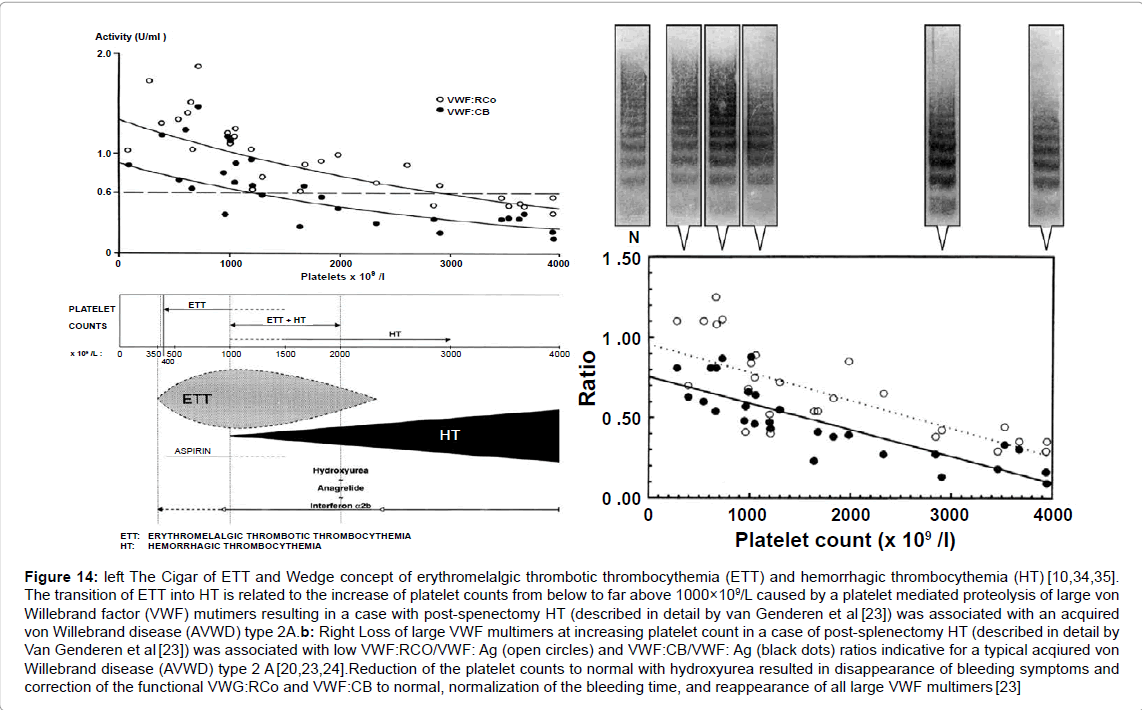
Figure 14: left The Cigar of ETT and Wedge concept of erythromelalgic thrombotic thrombocythemia (ETT) and hemorrhagic thrombocythemia (HT) [10,34,35]. The transition of ETT into HT is related to the increase of platelet counts from below to far above 1000×109/L caused by a platelet mediated proteolysis of large von Willebrand factor (VWF) mutimers resulting in a case with post-spenectomy HT (described in detail by van Genderen et al [23]) was associated with an acquired von Willebrand disease (AVWD) type 2A.b: Right Loss of large VWF multimers at increasing platelet count in a case of post-splenectomy HT (described in detail by Van Genderen et al [23]) was associated with low VWF:RCO/VWF: Ag (open circles) and VWF:CB/VWF: Ag (black dots) ratios indicative for a typical acqiured von Willebrand disease (AVWD) type 2 A [20,23,24].Reduction of the platelet counts to normal with hydroxyurea resulted in disappearance of bleeding symptoms and correction of the functional VWG:RCo and VWF:CB to normal, normalization of the bleeding time, and reappearance of all large VWF multimers [23]
The therapeutic implications from the prospective Rotterdam ET studies are completely in line with the concept that microcirculatory thrombotic complications in thrombocythemia already occur at platelet counts in excess of 400x109/L, which are relieved by reduction of platelet counts to normal (<400x109/L) or by control of platelet function with low dose aspirin (Figures 14 and 15). Thrombocythemia with platelet counts in excess of 1000+250x109/L are frequently associated with the paradoxical occurrence of thrombosis and bleeding. Mucocutaneous bleedings spontaneously occur at platelet count in excess of 1000 +250x109/L due to an acquired type II-like von Willebrand syndrome (AVWS, absence of high and intermediate von Willebrand factor (VWF) multimers) increasing in severity at increasing platelet counts to high levels above 1000 to 1500x109/L (Figures 14 and 15). Reduction of platelet count by anagrelide, interferon (or hydroxyurea if anagrelide and interferon fail) to less than 1000x109/L will result in the persistence of microvascular thrombotic events and the disappearance of the bleeding symptoms and AVWS (Figures 14 and 15). Correction of the platelet counts to normal is associated with no recurrences of microvascularevents and complete correction of the VWF-multimeric pattern and correction of all VWF-parameters to complete normal values. The study of Michiels et al. [3-11,31-34] and Van Genderen et al. [23-27,35-57] served as the scientific rationale for the ECLAP (European Collaboration on Low-dose Aspirin in PV) study design (1997) [33]. The ECLAP study nicely confirmed our concept of the effectiveness and safety of low dose aspirin in ET and PV in particular [38,39]. One low dose aspirin reduces the risk of microvasular ischemic or thrombotic complications from above 50% to less than 5% during 6.2 years of follow-up, and thereby prevented the progression from low thrombotic risk to high risk thrombotic in ET and PV [38,39].
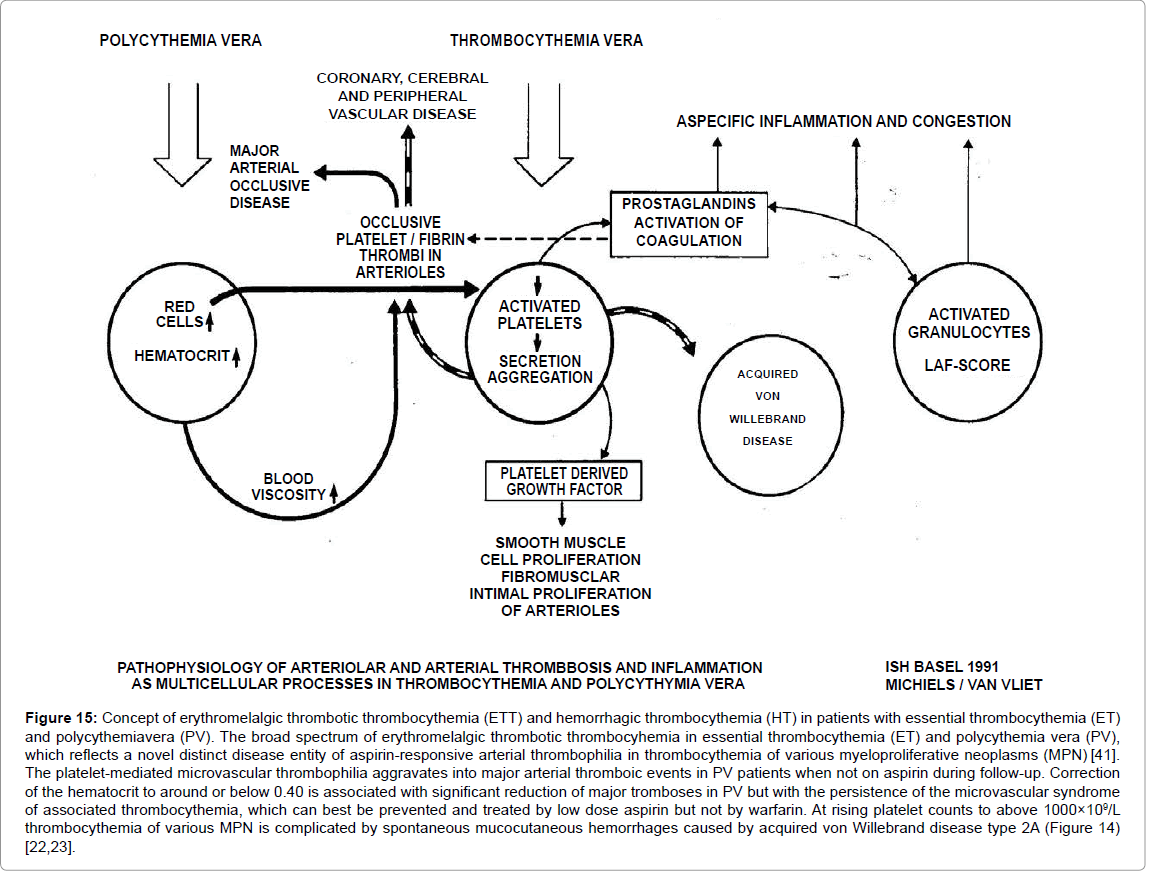
Figure 15: Concept of erythromelalgic thrombotic thrombocythemia (ETT) and hemorrhagic thrombocythemia (HT) in patients with essential thrombocythemia (ET) and polycythemiavera (PV). The broad spectrum of erythromelalgic thrombotic thrombocyhemia in essential thrombocythemia (ET) and polycythemia vera (PV), which reflects a novel distinct disease entity of aspirin-responsive arterial thrombophilia in thrombocythemia of various myeloproliferative neoplasms (MPN) [41]. The platelet-mediated microvascular thrombophilia aggravates into major arterial thromboic events in PV patients when not on aspirin during follow-up. Correction of the hematocrit to around or below 0.40 is associated with significant reduction of major tromboses in PV but with the persistence of the microvascular syndrome of associated thrombocythemia, which can best be prevented and treated by low dose aspirin but not by warfarin. At rising platelet counts to above 1000×109/L thrombocythemia of various MPN is complicated by spontaneous mucocutaneous hemorrhages caused by acquired von Willebrand disease type 2A (Figure 14) [22,23].
Low, Intermediate and High Thrombotic Risk in ET and PV Not on Aspirin
The risk stratification for thrombosis as low, intermediate and high thrombotic risk by Cortelazzo et al. in 199040 has been derived from a historical cohort of 100 ET patients not treated with aspirin thereby overlooking the original and updated key references of Michiels et al. [3,4,11,31-38,41] on the demonstration of aspirin responsive plateletmediated arteriolar and arterial thrombotic tendency inherent to thrombocythemia in ET and PV patients.
The characteristics of the thrombotic events in the retrospective Bergamo cohort of 100 patients were the following:
The age distribution of this cohort of ET did not reflect real life experience since the number of young ET patients was artificially manipulated to one third in the young age group to reach statistical significance. The risk for thrombotic complication was artificially low (1.7%) in thrombocythemia at young age below 40 years, but was high at age of >60 years (15%) not on aspirin, and moderately increased (6.3%) in the age group of 40 to 60 years, which is not significantly different from the thrombotic risk of 15% in the age group of >60 years [40]. Please note that in the Dutch prospective ET and PV studies the risk of thrombosis is untreated ET and PV is not age dependent and causally related to platelet-mediated thrombosis in the various stages of prefibrotic and early fibrotic MPN disease of ET and PV patients [36,37,40]. The type and number of 25 arterial and 3 venous thrombotic episodes in 20 out of a historical cohort of 100 untreated ET patients in the 1990 Bergamo study were mainly microcirculatory events including digital ischemia, transient ischemic attacks, superficial thrombophlebitis, ususual site of DVT, no stroke, and major thrombosis only in 4, myocardial infarction in 3 and femoral DVT in 1 (Table 6) [40]. It has been demonstrated at that time that such microcirculatory disturbances, superficial thrombophlebitis and TIAs are highly sensitive to low dose aspirin but not to coumarin [2]. If left untreated symptomatic ET patients with microcirculatory disturbances including erythromelalgia and atypical TIAs or visual symptoms are supposed to be at very high risk for digital ischemia, TIAs, stroke or acute coronary ischemic syndromes [1-7]. We concluded in 1995 [36] and 1997 [37] that the stratification in low, intermediate and high thrombotic risk in the 1990 retrospective Bergamo study40 has been derived from ET patients not on aspirin. The microvascular and major thrombotic complications in MPN-T disease of ET and PV patients at time of presentation and during follow-up while not on aspirin is independent of the degree of thrombocytosis or MPN disease burden in terms of splenomegaly, leukocytosis and constitutional symptoms. This means that the stratification in low, intermediate and high thrombotic risk in the retrospective Bergamo ET study is based on statistic misinterpretation and mystification leading to authorative overtreatment recommendation with hydroxyurea for MPN disease in ET and PV patients 42. The so-called high thrombotic risk ET as defined by a history or presentation of thrombosis at time of diagnosis or by reaching the age 60 years is not in line with the real incidence of thrombotic risk of regularly treated according to good clinical practice in the Rotterdam ET and PV studies at that time and beyond up to 2013 [11]. In our analysis and experiences the high thrombotic risk and its persistance in the 2012 International Prognostic Score for ET (IPSET) [42] with the indication to treat ET disease with hydroxyurea when reaching the age of 60 years alone irrespective of the degree of MPN disease burden is not appicable anymore as targeted 2014 guideline for ET and PV patients on aspirin with a low or intermediate MPN disease burden. Clinicians should realise that high thrombotic risk in the 2012 IPSET guidelines result in overtreatment of ET and PV patients who have low or intermediate MPN disease burden.
| Age | number of patients | Patient/years | Events number | Events % pt/yrs |
| 3411821.70% | ||||
| 40-60 years | 37 | 112 | 7 | 6.30% |
| >60 years | 29 | 73 | 11 | 15% |
| Total events in 20 of 100 ET patients=20% | ||||
| Cortelazzo et al. 1990 | number of patients | number of events | ||
| Total | 20 | 32 | ||
| Arterial | 17 | 25 | ||
| Micro: | digital ischemia | 7 | ||
| transient ischemic attacks | 15 | |||
| Major: | stroke | 0 | ||
| myocardial infarction | 3 | |||
| Venous | superficial | 3 | 7 | |
| thrombophlebitis | 3 | |||
| femoral DVT | 1 | |||
| Unusual localization DVT | 3 | |||
| Bleeding complications | 4 | |||
Table 6: The type and number of microvascular thrombotic events in the 1990
Bergamo study are very characteristic for untreated thrombocythemia [3].
The 1995 Bergamo study is a prospective randomized clinical trial (RCT) of 114 ET patients comparing hydroxyurea vs placebo in so-called high thrombotic risk ET patient [43]. This RCT is unbalanced since only 69% of the placebo group and 70% of the HU-treated ET patients received antiplatelet drugs, aspirin (effective in thrombocythemia [3,4] or ticlopedine (not effective in thrombocythemia [3,4]. The results show that 2 of 56 high thrombotic risk ET patients on hydroxyurea had major thrombotic events (one stroke, one myocardial infarction) and that 14 of 58 high thrombotic risk ET patients in the placebo group had mainly minor thrombotic complications including microcirculatory disturbances in 12, and major thrombosis in 2 similar as in the HU arm (MI 1, and deep vein thrombosis in 1) [43]. Moreover, 10 of the 14 symptomatic patients in the placebo arm manifested mainly microvascular disturbances and were not on treatment with aspirin [43]. A direct comparison of HU reaching normal platelet counts vs. low dose aspirin alone in ET patients at platelet counts below or around 1000×109/L is predicted to be equally effective for the prevention of microvascular circulation disturbances in ET (Figure 14 and 15). In the 1995 Bergamo study, the use of aspirin in the placebo arms as compared to the HU arm was not associated with increased bleeding events at platelet counts around 1000×109/L [35]. The recommendation from this unbalanced RTC (HU vs placebo) to use hydroxyurea in so-called high thrombotic risk ET as first line treatment for ET is not justified and potentially leukemogenic and has led to significant world-wide over-treatment of ET patient who usually have low to intermediate MPN disease burden and a normal life expectance [43].
The combined low dose aspirin platelet reduction strategy to lower platelet count to about values around 600x1012/L (near normal platelet concept) in symptomatic ET-patients by anagrelide or, interferon (hydroxyurea when anagrelide and interferon failed) is most effective in daily practices for the treatment of thrombocythemia in ET and PV [11]. Prospective studies comparing this concept of near normal platelet count strategy versus low dose aspirin alone is warranted. The prediction is that the near normal platelet count strategy in symptomatic JAK2 mutated ET, and ET mimicking PV (prodromal PV) seems to be far superior to low dose aspirin alone, simple because low dose aspirin will significantly increase the risk of bleeding complications at platelet counts around and above 1000 x 109/L (Figures 14 and 15).
Thrombosis and Bleeding Risk in JAK2 Mutated ET and PV and JAK2 Wild Type ET
With the advent of molecular screening of MPN patients it should be realized that WHO-ET patients with a JAK2V617F mutation load of less than 50% of the granulocytes are indeed heterozygous for the JAK2 mutationl [44-46]. However, WHO-PV patients with a JAK2V617F mutation load of less than 50% of the granulocytes proved to be combined heterozygous homozygous positive for the JAK2V617F mutation [44-46]. Vannucchi et al determined the JAK2V617F mutation allele burden as expressed in percentages of JAK2V6717F mutated granulocytes of 173 PV patients at diagnosis47. The JAK2V617F allele burden in granulocytes ranged from 1-25% in 33%, from 25-50% in 29%, from 50 to 75% in 20% and from 75 to 10% in 18% and treatment consisted of phlebotomy in 49% and cytoreductive therapy (mainly hydroxyurea) in 51% [47]. The JAK2V617F allele mutation burden correlated with MPN disease activity in terms of stimulated erythropoiesis by higher hematocrit and erythrocytes, lower MCV, serum EPO and ferritine, and stimulated myelopiesis by higher leukocytes, serum LDH and LAP score [47]. Comparing by statistics alone the PV patients with low JAK2V617F (1 to 50%) versus high JAK2V617 (50-100%) allele burden, the relative risks (RR) for MPN disease burden increased from 1 to 4 for pruritis, from 1 to 4 for palpable splenomegaly and from 1 to 4 for spleen sizes above 15 cm length diameter on scan [47].
In a subsequent retrospective study Vannucchi et al assessed in 2007 the incidence of thrombosis related to the JAK2 allele burden in a large retrospective study of 962 MPN-T patients subdivided in 323 PV and 639 ET patients [48]. Aspirin responsive platelet thrombophilia or microvascular symptoms due to microvessel disorder including migraine-like headache, acral paresthesia, erythromelalgia, transient neurological and visual disturbances (Sticky Platelet Syndome 49) were excluded by definition and not considered in this retrospective analysis [48]. Major thrombotic events were assessed as could be objectively documented in the medical records including ischemic stroke, transient ischemic attacks, myocardial infarction, angina pectoris, deep vein thrombosis abdominal vein thrombosis, and pulmonary embolism occuring at diagnosis or follow-up when not on aspirin48. According to these exclusion and inclusion criteria, the clinical profile and the incidence of major thrombotic events in 188 JAK2V617F homozygous MPN patients (JAK2V617F mutation above 50% in 104 PV and 14 ET) and in 587 heterozygous (JAK2V617F mutation less than 50% in 219 PV and 257 ET) and 257 wild type ET patients was assessed and calculated in Table 7 and Figure 16. Homozygous JAK2V617F positive patients with an allele mutation load above 50% were older, had higher leukocyte counts, hematocrits and larger spleen volumes indicating advanced MPN disease [48]. One hundred seventy-six patients (18.3%) had a major thrombotic event at diagnosis with a similar frequency in PV (19.2%) and ET (17.8%). A similar incidence was found in our analysis of the literature in 1241 ET patients not on aspirin from 14 retrospective studies 50. During long-term follow-up, major thrombosis (usually not on aspirin) occurred in 122 patients (12.7%), corresponding to 14.9% in PV and 11.6% in ET patients and hemorrhages at diagnosis manifested in 55 (5.7%) patients, 5.3% in PV and 6.0% in ET20. The overall incidence of major thrombotic events during 10 years followup usually not on aspirin was about 20% in ET heterozygous for the JAK2V617F mutation and in about 10% for JAK2 wild type ET [48]. A hemorrhage during follow-up was recorded in 45 (4.7%) ET/PV patients. A similar incidence of hemorrhages was found in our analysis of the literature in 1241 ET patients from 14 retrospective studies [50]. The frequency of bleeding was higher in JAK2V617F homozygous (21.4%) than in wild type or heterozygous ET patients, 3.1% and 3.8% respectively. The higher bleeding tendency in homozygous JAK2V617F MPN patients is predicted to be related to higher erythrocyte counts at increased platelet and leukocyte counts and its pathophysiology of the underlying mechanisms is currently under our investigation.
| Patients | PV N=323 | ET N=639 | ||
| JAK2V617 mutation status | hetero | homozygous | hetero | wild type |
| Number of patients | 219 | 104 | 368 | 257 |
| At diagnosis | ||||
| Major arterial events | 21% | 15,4% | 21.70% | 10.50% |
| Venous events | 6.40% | 2.90% | 7.90% | 4.70% |
| During 10 years follow-up (not on aspirin) | ||||
| Major arterial events | 10.10% | 12.50% | 6.30% | 5.80% |
| Venous events | 4.10% | 7.70% | 6.30% | 2.70% |
| Total during life time follow-up | ||||
| Major arterial | 31.10% | 27.90% | 28% | 16.30% |
| Venous | 10.50% | 10.60% | 14.20% | 7.40% |
Table 7: Major cardiovascular events at diagnosis or during long-term follow-up in 323 PV and 639 ET patients according to the JAK2V617F mutation status in the retrospective study of Vannucchi [48]. Only major thrombotic events were retrospectively recorded not including the erythromelalgic peripheral and migraine like cerebral ischemic events [48]. In 14 homozygous ET patients total major arterial and venous events occurred in 78.6% and 57.1% respectively.
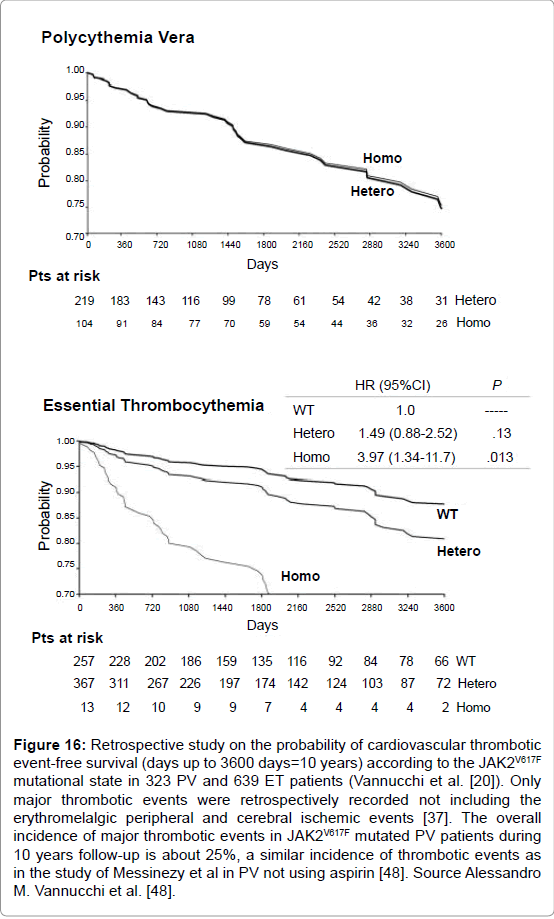
Figure 16: Retrospective study on the probability of cardiovascular thrombotic event-free survival (days up to 3600 days=10 years) according to the JAK2V617F mutational state in 323 PV and 639 ET patients (Vannucchi et al. [20]). Only major thrombotic events were retrospectively recorded not including the erythromelalgic peripheral and cerebral ischemic events [37]. The overall incidence of major thrombotic events in JAK2V617F mutated PV patients during 10 years follow-up is about 25%, a similar incidence of thrombotic events as in the study of Messinezy et al in PV not using aspirin [48]. Source Alessandro M. Vannucchi et al. [48].
In the second Vannucchi study, a total of 214 patients were treated with phlebotomy, 58% of 219 PV and 4% of 257 ET patients. Myelosuppressive chemotherapy was administered to 497 patients (52%) including 59% of 219 PV and 48% of 257 ET patients [48]. For comparison, in the Dutch 2008 survey of 363 MPN (123 ET, 190 PV and 50 MF) patients 93% of PV, 71% of ET and 37% of MF were on aspirin mainly because of microvascular symptoms including migraine-like headache, acral paresthesia, erythromelalgia, transient neurological and visual disturbances. Phlebotomy became the first line treatment in 6% of ET, 78% of PV and 9% of MF when the Dutch recommendations are applied [51,52].
Because of advanced or symptomatic MPN disease 31% of ET, 29% of PV and 30% of MF were on treatment with hydroxyurea and 16% of ET, 16% of PV and 4% of MF were on treatment with pegylated interferon (PegasysR) [11,54,55]. The 20% difference of HU use (50% of Italian MPN-T patients versus 30% of Dutch MPN patients) can readily be ascribed to significant differences in the Italian IPSET recommendations 42 versus the Dutch guidelines for MPN disease in ET and PV patients [50-54].
Hydroxyurea is Not an Innocent Myelosppresive Agent in PV and ET
Hydroxyurea is not an innocent drug and should be used with caution and withheld as long as phlebotomy and low dose aspirin are effective in the treatment of early and intermediate plethoric PV stages 1 and 2. In the 1980 French PV study in PV patients under the age of 65 years, toxicity of hydroxyurea was observed within 5 years in 29% of 133 PV patients, which was limited to dry skin and acne in 7%, gastric pairddiarrhea in 9%, aphthous ulcers in the mouth in lo%, and leg ulcers in 9% [56]. Leg ulcers only healed after discontinuation of hydroxyurea; these complications generally appear late within 5 years or more after initial treatment. Dry skin in 1, aphthous stomatitis in 4 and leg ulcers in 10 cases were the reasons for replacing hydroxyurea by pipobroman in 9%. In the update 10 years later in 1997, the incidence of leukemia was about 10% at 13 years in hydroxyurea treated PVpatients and life expectancy was 70% at 14 years as compared to 83.7% in age-matched controls [56]. The frequency of MF or spent phase PV in the update in 1997 was 17% at 10 years and 40% at 16 years. The final analysis of this 1980 French PVSG study of HU as the upfront first-line therapy in 136 evaluable PV patients younger than 65 years is published in 2011 [57]. The cumulative incidence (probability) of myelofibrosis (MF) at 10, 15 and 20 years was 15%, 24% and 32% in the HU arm and the cumulative incidence of AML/MDS at 10, 15 and 20 years was 7.3, 10.7% and 16.6% for HU treated PV patients. As shown in Table 8, high risk PV with advanced MPN-T disease in ET and PV patients in terms of high JAK2V617F allele burden, progressive MPN disease, splenomegaly and constitutional symptoms are candidates for myelosuppressive (hydroxyurea) or myeloreductive (JAK2 inhibtors) treatment [11,41].
| PV: WHO-ECMP stage | 0 | 1 | 2 | 3 | 4 | 5 | 6 |
| WHO-ECMP | Prodromal masked PV | Erythrocy-themic PV | Early PV | Overt PV | PV PMF | Post-PV MF | Leukemic PV |
| Clinical Diagnosis | Classical PV | Masked PV | Spent PV | MDS/AL | |||
| LAP-score | ↑ | ↑ | ↑ | ↑ | ↑/↑↑ | variable | variable |
| Red cell mass (RCM) | N | ↑ | ↑ | ↑ | ↑ | variable | N/↓ |
| Serum EPO | N/↓ | N/↓ | ↓ | ↓ | ↓ | variable | N/↓ |
| Erythrocytes x1012/l | >5.8 | <5.8 | >5.8 | >5.8 | >5.8 | variable | N/↓ |
| Leukocytes x109/L | <12 | <12 | <15 | < or->15 | >15 | >20 | >20 |
| Platelets x109/L | >400 | ,400 | < or >400 | >400 | < or >1000 | variable | variable |
| WHO-ECMP bone marrow | Early PV | Early PV | Early PV | Trilinear PV | Trilinear PV | Myelofibrosis | Leukemic |
| Bone marrow cellularity (%) | 50-80 | 50-80 | 60-100 | 80-100 | 80-100 | Decreased | Increased |
| Grading reticulin fibrosis: RF | RF 0-1 | RF 0-1 | RF 0-1 | RF 0/1, | RCF 2/3 | RCF 3/ 4 | |
| Grading myelofibrosis: MF57 | MF 0 | MF 0 | MF 0 | MF 0 | MF 1/2 | MF 2/3 | |
| Splenomegaly on palpation | No/+ | No | No/+ | + | ++/+++ | /large | large |
| Spleen size, echogram cm | <12-15 | <13 | 15-Dec | 18-Dec | 18->20 | >20 | >20 |
| Spleen size on palpation cm | 0-3 | NP | 0-3 | 6-Apr | >6 | >8 | >8 |
| JAK2V617F in Granulocytes % | low | low | Moderate <50 | High >50 | High >50 | High >50 | No or ++ |
| JAK2V617F in BFU-e (exon 12) | +(++) | +(++) | +(++) | ++ | ++ | ++ | |
| Therapeutic implications | Low risk | Low risk | Low risk | Intermediate risk PV | High risk | Post-PV MF | Leukemia |
| Anno 2014 | MPN | MPN | MPN | PV-MF | Spent phase PV | ||
| First line Aspirin/Phlebotomy | Aspirin | Aspirin | Phlebotomy | Phlebotomy* | If IFN resistant à | JAK2 | Chemotherapy |
| Second line IFN versus | Phlebotomy | Phlebotomy | Aspirin | Aspirin | HU or JAK2 | Inhibitor à | Bone marrow transplantation? |
| Hydroxyurea (HU) | Low dose IFN à responsive | IFN à resistant à HU | inhibitor | Bone marrow | Supportive | ||
| Third line JAK2 inhibitor | transplantation |
Table 8: Staging of JAK2V617F positive prodromal PV, erythrocythemic PV, and five stages of PV according to WHO-ECMP criteria related to therapy anno 2013 [41] *↑ = increased, ↓ = decreased, N = normal, + = present or heterozygous; ++ = homozygous.