Enzyme Engineering
Open Access
ISSN: 2329-6674
ISSN: 2329-6674
Mini Review - (2015) Volume 4, Issue 1
Use of Computational (in silico) methods are widely applied in drug discovery. In drug discovery process, identification of the suitable drug target is the first and foremost task. These targets are biomolecules which mainly include DNA, RNA and proteins (such as receptors, transporters, enzymes and ion channels). Validation of such targets is necessary to exhibit a sufficient level of ‘confidence’ and to know their pharmacological relevance to the disease under investigation. The aim of this mini-review is to illustrate some of the in silico methods that are used in drug discovery, and to describe the applications of these computational methods.
The drug discovery process involves the identification of the lead structure followed by the synthesis of its analogs, their screening to get candidate molecules for drug development [1]. The goal of the drug discovery process is to search for new drug molecules which can bind to a specific target known to be involved in causing a disease and change the target’s function [2-4].
In the traditional drug discovery process, identification of the suitable drug target is the first and foremost task [5]. These targets are biomolecules which mainly include DNA, RNA and proteins (such as receptors, transporters, enzymes and ion channels). Validation of such targets is necessary to exhibit a sufficient level of ‘confidence’ and to know their pharmacological relevance to the disease under investigation [6]. This can be performed from very basic levels such as cellular, molecular levels to whole animal level. Once the target validation has performed, effective compounds such as inhibitors, modulators or antagonists for such target need to be identified. This process is called lead identification where the design and development of a suitable assay is done to monitor the effect on the target under study [7]. High-Throughput Screening (HTS) plays a crucial role in this phase where large numbers of chemical compounds are exposed to the target. Compounds showing dose-dependent target modulation in terms of a certain degree of confidence are processed further as lead compounds. Subsequently, the experiments are performed on the animal models in the laboratories and the positive results are then optimized in terms of potency and selectivity. Physicochemical properties and their pharmacokinetic and safety features are also assessed before they become candidates for drug development [8]. Even though most of the processes depend on experimental tasks, in silico approaches are playing important roles in every stage of this drug discovery pipeline (Figure 1).
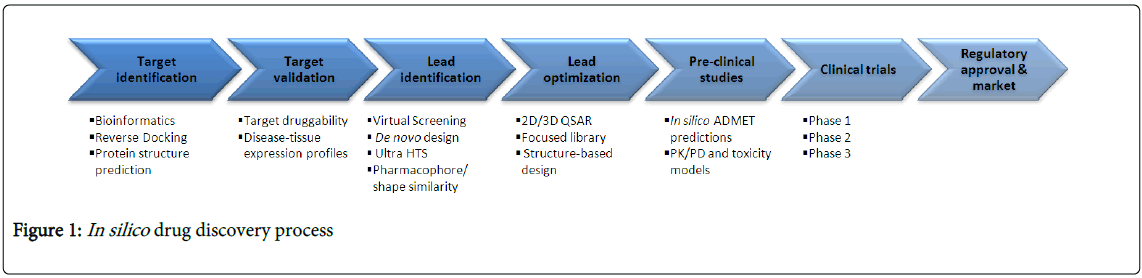
Figure 1: In silico drug discovery process
Drug discovery and development process
The important steps involved in the process of drug discovery are:
Lead identification
A critical factor is the search for lead structures. Leads can be obtained from natural products, structure directed molecular design, modification of natural products, biochemical understanding of the disease process and broad screening of synthetic compounds etc.
Lead optimization
It involves the identification of lead molecule through the synthesis and testing of derivatives of leads to develop Structure Activity Relationships (SARs), calculation of physico-chemical properties and using them for lead refinement from techniques like Quantitative Structure Activity Relationships (QSARs). Modern techniques such as combinatorial chemistry coupled with High Throughput Screening (HTS) provide us an enormous number of New Chemical Entities (NCEs) but these techniques have not been proved cost effective due to high costs of reagents in addition to the involvement of costly modern equipments, hence there is a strong demand for the computer assisted techniques which are fast, reliable and cheap to rationalize these early steps in drug development [9].
Pre-clinical lead development
Drug formulation experiments, in-vivo studies in animals, animal safety studies, drug metabolism studies and large scale synthesis come under the umbrella of pre-clinical lead development [10].
Clinical lead development
It involves the small scale safety and dose ranging test in healthy human volunteers (phase I) and development of clinical study protocols employing clinical investigations on patients (phase II) and comparative double blind studies on patients’ studies (phase III).
Several sequence and structure-based approaches have been proposed for target identification. In the sequence-based method, functional information about the target and its positioning in the biological networks is provided to detect unique targets from the disease causing pathogens (e.g. bacteria or viruses) by comparing functional genomics of humans with corresponding genomics of pathogens [11].
The general methodology followed for finding a novel targetspecific- drug for a given disorder involves random high-throughput screening (HTS) [8]. In this process, large chemical-libraries are checked for their capacity to block and/or modify the target. For instance, if the target is a novel beta lactamase enzyme, compounds will be screened for their ability to enhance or reduce the activity of that receptor or altogether block it [12]. In this manner, novel pharmacophores are being discovered at a significant pace nowadays.
Furthermore, an important aspect of such screenings is that the selectivity of the compounds for the target is also deduced by concurrently screening the hits against other targets. This is called as cross-screening. The significance of cross-screening lies in the fact, that in case the compound hits frequently at non-related targets; it is expected to cause significant ‘toxicity’ in the human body (in case it reaches to the stage of human clinical trials) [3].
At this juncture, it is noteworthy to mention about another screening method referred to as Virtual High Throughput Screening (vHTS). In this type of screening method, computer-generated models are used. These computer programs attempt to dock virtual libraries consisting of 3-D structures of chemicals to a target. This method is quick, reliable and economic as well [13-15].
Through these methods the scientists discover ‘a lead molecule series’. The chemical structures included in this series are expected to possess adequate target specificity and favorable properties for ‘druglikeness’. Following this, one or two compounds may then be selected for drug development. The best compound of these is termed as the "lead" compound [14,15]. The lead compound is further optimized by random trials or more judiciously by rational drug design methods. The leads are examined for whether these compounds share some common features, chemists/modelers then use Structure-Activity Relationships (SARs) to improve certain structural features of the lead molecules in order to (i) increase activity against the chosen target, (ii) reduce activity against unrelated targets, and (iii) improve the "druglike" or ADME properties of the molecule.
A major target in the drug discovery process is to develop a high degree of “rationality.” This would represent an intellectually rigorous approach incorporating Computer-Assisted Molecular Design (CAMD), limited but highly focused chemical synthetic effort, and sophisticated biological assays. Thus, the drug design process may become significantly less risky and more resource efficient [13].
The rational drug design approaches are maturing in the sense that they are moving from being only a theoretical promise based on their use in the retrospective analysis of known compounds, to innovative design and discovery of Novel Chemical Entities (NCEs) and providing solutions to novel problems. There are a number of successful examples in the literature using rational design approaches for design and discovery of NCEs [16-18]. The advances in computer hardware and software have made this process more effective and time efficient. The systematic evolution of drug discovery research is outlined in Table 1.
| Time | Materials | Test systems |
|---|---|---|
| Ancient time | Plants, venoms, minerals (Natural Products) | Humans |
| -1806 | Morphine | |
| - 1850 | Chemicals | |
| - 1890 | synthetics, dyes | animals |
| - 1920 | synthetics, dyes | animals, isolated organs |
| - 1970 | synthetics, dyes | enzymes, membranes |
| - 1990 | Combinatorial libraries | Human proteins, HTS |
| - 2000 onwards | Focused libraries | CAMD, vHTS, virtual screening |
Table 1: The Evolution of Drug Research
In silico techniques significantly contribute to early drug discovery and are important in target and lead discovery. It can be anticipated that the contribution of in silico techniques in drug discovery will increase substantially in the future. Ultimately, to have a much broader impact, the use of in silico methods will need to become a part of every researcher involved in drug discovery. The need for timely adaptation and application of in silico approaches in pharmaceutical research has clearly been recognized and is expected to improve further the overall efficiency of drug discovery.