Drug Designing: Open Access
Open Access
ISSN: 2169-0138
ISSN: 2169-0138
Research Article - (2021)Volume 10, Issue 4
Design of a drug compound that can effectively bind to the E channel and block the diffusion of hydrogen (H+) and K+ ions through channel and inhibit virus life cycle and replication is an important task. A new class of positively charged, +2 e.u., molecules is proposed here to block proton diffusion trough the E channel. Several drug candidates, derivatives of a lead compound (diazabicyclooctane), were proposed and investigated drug binding energy and positions. E protein has in-channe l and out of channelbinding sites of high affinity for suggested molecular blockers. The most promising structure of E channel blocker is suggested.
SARS-cov-2; E channel inhibitors; Molecular dynamics; Ligand binding
The COVID-19 viral pneumonia pandemic coerced by SARS-cov- 2 virus has swept across all countries at the beginning of 2020 in one year has already claimed more than 2 million lives (https://COVID-19.who.int, 2021). Therefore, a wide population vaccination and development of effective treatment of the decease are necessary. The paper suggest drug molecules, which are able to bind effectively with the membrane channel formed by E protein and disrupt it’s right functioning and the SARS-cov-2 virus in whole. The development of an effective drug is a task of great importance because of the ability of SARS-cov-2 virus to infect fast a large population, sometimes probably through cross-species barriers, and to mutate quickly.
A reasonable approach to affect and disrupt the functional cycle of the SARS-cov-2 virus particle, (Figure 1), is to specifically affect the envelope protein E, which forms the trans-membrane channel E, an important element that ensures its proper functioning. The E channel is formed by amino acid residues 8-65 of each of the five subunits A, B, C, D, E. The five interacting α-helices build up the E protein (Figure 2), which has central channel of large diameter ~17-19 Å, if measured between Cα atoms of the NMR structure, PDB code 5X29 solved at T=308K, pH=5.5, 50 mM NaCl, [1]. The channel activity of the E protein of the SARS-cov-2 coronavirus was shown to be critical for infection process of living cell. For example, pathogenicity was significantly hampered when the transmembrane segment of infectious bronchitis virus E protein was replaced with a heterologous domain that lacked ion channel activity [2].

Figure 1: SARS-cov-2 virus, E (envelop) protein forming E channel.

Figure 2: NMR structures of E protein, T=308K, pH=5.5, 50 mM NaCl. Sixteen variants reflect thermal dynamics of E protein structure.
The importance of ion channel functionality for virulence has been demonstrated in several viruses. For example, the H+ channel activity of the M2 protein of influenza A is critical for infection in two ways: 1) following endocytosis, the channel facilitates H+ [3] and K+ transport into the viral lumen, enabling viral RNA to dissociate and initiate replication [4]. In corona viruses, the channel activity of the E protein was shown to be critical for infectivity. It is shown that virus pathogenicity was significantly reduced when the transmembrane segment of infectious bronchitis virus E protein was replaced with a heterologous domain that lacked ion channel activity [2]. In SARS-CoV-1 virus, studies have shown that viruses in which the channel activity of the E protein was abolished, were far less infectious [5,6]. A recent computational study of interaction of SARS-CoV-2 virus with several drug molecules affecting replication cycle [7] suggest molecular structures to inhibit the process. The structure of coronavirus protein E is the least understood in terms of mechanism of action and structure. Functionally, the E protein has been implicated in viral assembly, release, and pathogenesis [8] and it is crucially that E proteins are important for viral pathogenesis [9]. The small size of E channel and overall hydrophobicity prompted suggestions that coronavirus E proteins might be functionally similar to viroporins. Indeed, E channel is small and usually hydrophobic multifunctional viral proteins that modify cellular membranes, thereby facilitating virus release from infected cells. E proteins from several coronaviruses: including SARS-CoV-1 [10,11], MERS coronavirus, human coronavirus 229E and mouse hepatitis virus [12,13], and infectious bronchitis virus [14], were shown that the cation selective channel activity may be blocked by Hexamethylene amiloride [14,15]. The importance of ion channel functionality for virulence has been demonstrated in several viruses. For example, the H+ channel activity of the M2 protein of influenza A is critical to infectivity in two ways: Following endocytosis, the channel facilitates H+ [3] and K+ transport [4] into the viral lumen, enabling viral RNA initiate replication. It is also shown that the E channel can be inhibited by Gliclazide and Memantine molecules [6].
Approaches to specifically affect the proton channel formed by the trans-membrane domain of the matrix protein M2-TMD of influenza A virus is well known. Proton diffusion through M2 channel can be suppressed via binding of a special blocker molecule inside the M2 ion channel that physically prevents proton diffusion through the channel. Recently it was shown that the M2 ion channel of influenza A virus can be effectively blocked by molecule 1,4-diazabicyclo[2.2.2] octane and the most effective blocker molecule has been constructed [15,16]. The successful design of effective drug molecules blocking the M2 ion channel of influenza A virus, motivated us to examine possibility of design of molecules to block the E protein ion channel of SARS-CoV-2 virus. The coronavirus E proteins from several coronaviruses were shown to posses channel functionality [11-13,17-19] and E proteins are important for viral virulence. Hence, any inhibitor, channelblocker, found against the SARS-CoV-2 E protein may serve as an anti-COVID-19 drug candidate. A recent paper [19] reports NMR structure of E protein channel which has a minimal diameter ~6 Å in the first half of the channel between residues N15 of five α-helixes are forming E channel. In our recent paper [15] we developed method to construct and investigate effectiveness of drug molecules to block M2 ion channel via blind docking and binding energy calculation of a set of drug molecules on the M2 channel of influenza A virus.
Here, we suggest a novel drug molecules which effectively binds to E protein ion channel and block proton transport through the channel and thereby affect a proper functioning of that channel. New set of antiviral compounds, derivatives of 1,4-diazabicyclo-[2.2.2]octane (DABCO) is proposed here. New blocker molecules effectively interact with the E channel of SARScov2 virus and should considerably affect a proper functioning of E channel and on that virus in hole.
Method of modeling
Atomic structure and temperature dynamics of E protein, at human body temperature, were modeled by using the fifteen PDB structures, (pdb code 5X29), which are representing thermal dynamics at temperature 308K and pH 5.5 [1], (Figure 2).
Docking of blocker molecules is done via hierarchical blind docking method hbDOCK, which is described in details else were [20] and consists of two stages: (i) exhaustive search and rough ranking of preliminary set of positions, and (ii) refinement for the set of fifteen experimental structures of E protein via simulated annealing method. The total binding energy ΔG(x) of drug to E protein in conformational state x, where x is a set of atomic coordinates, is calculated as the sum of terms,
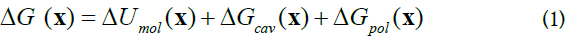
where for a set of atomic coordinates x, i.e. for all atomic positions ri for a set of atoms of protein-drug complex, Umol(x) energy of all pair atom-atom interactions, i.e. bond and valence angle deformations, torsional and improper angle deformations, van der Waals and electrostatic pair interactions and hydrogen bond; Gcav(x) molecular cavity formation, Gpol(x) energy of polarization of out of protein volume environment. All atom-atom interaction are calculated with AMBER94 and AMBER-GAFF [21-23] force field. Energy Gcav(x) is calculated as,

where, γ = 39 cal/mol/Å, S(x) is the molecular surface (MS) area, which is calculated by the SIMS method [24,25]. The solvent polarization energy Gpol(x) is calculated via Generalized Born method with optimized Generalized-Born, NSR6 method [26,27] using additional minor, ~5%, correction [25-29],
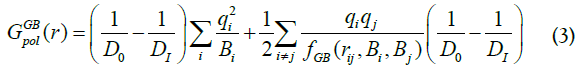
where Di is the internal molecular dielectric constant, Di=12; D0 is the dielectric constant of the external volume, D0=30; qi is atomic charge; rij interatomic distance, Bi is Born radii. Function fGB, [26], is,
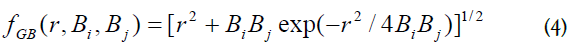
Born radii Bi is defined as integral over MS surface,
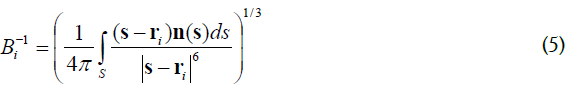
where n(s) is a normal vector to the MS at the point s. The optimized GB-MSR6c estimates solvent polatization energy with errors ~ 2.5% compare to the most accurate Poisson-Boltzmann [26-29].
The exhaustive docking procedure [20] consists of several steps: (i) a search for all possible binding sites on the protein molecular surface accessible to the drug molecule, (ii) selection of potential binding sites by rank, (iii) global optimization for ligand position/orientation and energy of binding using MD simulated annealing method with the set of 72 initial ligand orientations. The set-72 consist of 12, icosahedron, unit vector orientations (φ, Θ) angles, with 6 ψ-angle orientations, with the step of π/3, for each of 12 (φ, Θ) pairs.
The protein forming E channel, PDB code 5X29, is embedded into the lipid cell membrane. Experimental measurement indicates that dielectric constant Din of the lipid membrane (i.e., outside E protein volume [24]), while Dout, dielectric constant of outside volume is equal to ~30. The E protein volume and lumen volume of the E ion channel itself are considered here as the internal protein volume with a dielectric constant Din of 12 to 16 [30-33].
Structure of E protein channel
The atomic structure of E protein has been determined by NMR method at T=308K, pH=5.5, 50 mM NaCl, PDB code 5X29 [1], (Figure 2). The PDB entry 5X29 consists of 16 structures, which reflect the thermal dynamics of E protein. The structure of the E channel obtained by NMR include all hydrogen atoms and therefore full information about the ionization state of ionizable residues. The E protein is unique in a sense that it has only one ionizable residue GLU8A (B, C, D, F) in each of five equivalent α-helices with pKa0=4.25. Therefore, the experimental NMR structure of E protein at pH=5.5 has ionized form of GLU8A (B, C, D, F) residues, so that the ionized state of these residues will be the same at physiological pH 6-8 units. Thermal dynamics of E protein, PDB 5X29, is presented by the 16 NMR models witch reflect conformational flexibility of E protein structure at temperature 308K, showing RMSD 3.4 Å (Figure 2).
Novel inhibitors of E channel
The 3D structure of the E protein channel consists of the pentamer of five equivalent α-helices, which form the molecular E channel, (Figure 2). Function of E channel consists of a pumping of H+ protons and K+ ion through the cell membrane, similar to the function of the M2 channel of influenza A virus [3,4]. Activity of the E channel can be considerably destroyed via putting a molecular “cork” inside the E channel, i.e. a specially designed molecule, which is able to effective binding inside the E channel and sterically and electrostatically prevent transport of positively charge cation through the channel, as it is shown for M channel of influenza A virus [15,16]. It is shown that the E channel can be inhibited by Gliclazide and Memantine molecules [6].
It should be noted that structure of coronavirus protein E is the least understood in terms of mechanism of action and structure. Functionally, the E protein has been implicated in viral assembly, release, and pathogenesis [8,9] and it is crucially that E proteins are important for viral pathogenesis [10-34].
A new class of molecular inhibitors of the E channel is based on diazabicyclooctane (DABCO), N2(C2H4)3 (Figure 3a), which serves as a lead compound [35-36]. Recently it was shown that molecule DABCO and its derivatives can effectively inhibit the proton transfer through the M2 ion channel of influenza A virus [15,16]. The current work suggests a set of new molecules, derivatives of the DABCO, (Figure 3a), and analyze their ability to be strongly bound inside the E channel of the SARS-cov-2 virus.

Figure 3(a): Structure of drug molecule DABCO.
Leading compound
Exhaustive docking has been performed on the molecular surface of the SARS-cov-2 molecule, modeled by NMR structure, PDB code 5X29. Protein molecular surface accessible for water molecules was analyzed via the HBDock technique [20]. It must be noted that the HBDock method implements the docking of a flexible drug molecule to a flexible protein molecule via a multiple simulated annealing approach and global optimization over rotational and translational degrees of freedom of a ligand molecule.
The result of the DABCO docking onto the E channel is illustrated in (Table 1). It can be seen, Table 1, that the both in-channel binding positions have large binding energies, -16.5 kcal/mole, large van der Waals, coulomb energies and forming two almost ideal H-bonds with energies~-5 kcal/mole each. There are the second in channel binding position with considerably lower binding energy, by ~6 kcal/mole, and negligible occupation rate. A blocking effect of proton diffusion through the E channel has basically electrostatic nature due to the positive charge, +2, of the DABCO molecule. Steric blockage is negligible for proton H+ even proton is solvated complex with six water molecules. It can be noted that the in-channel binding mode with energy -16.5 kcal/ mole has the energy gap, EGAP=EOUT × EIN ~3 kcal/mole, i.e. ratio of fractions of IN and OUT channel bound molecules is negligible (CIN/COUT) ~0.01 (Table 1).
| Drug molecule | ePLb | eVDWc | eCould | eHbe | XHf∙∙∙Yg/a.a. | Dist, Å | in(out)i |
|---|---|---|---|---|---|---|---|
| DABCOa | -16.5 | -8.1 | -2.9 | -9.9 | GLU8A-OE2∙∙∙HN1 | 1.91 | In |
| GLU8E-OE1∙∙∙HN2 | 2.00 | ||||||
| -13.4 | -10.2 | -0.3 | -9.8 | VAL24C-O∙∙∙HN2 | 2.09 | out | |
| LEUE65-OXT∙∙∙HN1 | 1.94 | ||||||
| -9.9 | -11.8 | 4.9 | -9.7 | OE1-GLU8A∙∙∙HN1 | 1.92 | In | |
| OD1-ASN15A∙∙∙HN2 | 2.06 | ||||||
| DABCOB | -25.2 | -1.8 | -7 | -9.9 | LEU9C-OXT∙∙∙HN2 | 1.97 | Out |
| VAL47C-O∙∙∙HN1 | 1.98 | ||||||
| -23 | -13.9 | -6.9 | -9.9 | LEU65B-O∙∙∙HN1 | 2.09 | Out | |
| VAL47C-O∙∙∙HN2 | 2.03 | ||||||
| -20.2 | -16.4 | -0.8 | -9.7 | ASN15C-OD1∙∙∙HN1 | 2.14 | in | |
| GLU8E-OE1∙∙∙HN2 | 2.08 | ||||||
| DABCO3B | -30.9 | -28.4 | -4.9 | -8.9 | ASN15C-OD1∙∙∙HN1 | 2.12 | in |
| GLU8E-OE1∙∙∙HN2 | 2.25 | ||||||
| -30.7 | -31 | -3.7 | -9.9 | LEU65A-OXT∙∙∙HN1 | 2.1 | out | |
| LEU21C-O∙∙∙HN2 | 2.18 | ||||||
| -28.4 | -28.6 | -5.3 | -9.6 | LEU65B-OXT∙∙∙HN1 | 2.01 | out | |
| VAL47C-O∙∙∙HN2 | 1.98 | ||||||
| DABCON | -24.7 | -16 | -3.2 | -9.7 | GLU8A-OE2∙∙∙HN1 | 2.06 | in |
| ASN15C-OD1∙∙∙HN2 | 2.14 | ||||||
| -23.3 | -17.3 | -4.4 | -9.9 | PHE20D-O∙∙∙HN1 | 2.07 | out | |
| LEU65C-OXT∙∙∙HN2 | 1.93 | ||||||
| -22.4 | -24.1 | 2.9 | -9.7 | SER50B-O∙∙∙HN1 | 2.12 | out | |
| TYNB-OH∙∙∙HN2 | 2.08 | ||||||
| DABCO3N | -34.7 | -34.1 | -6.8 | -9.1 | ASN15C-OD1∙∙∙HN1 | 2.19 | in |
| GLU8E-OE1∙∙∙HN2 | 2.15 | ||||||
| -30.9 | -38.1 | -3.4 | -5 | LRU65C-OXT∙∙∙HN1 | 2.01 | out | |
| -30.8 | -40.1 | -0.2 | -4.8 | GLU8A-OE1∙∙∙HN1 | 2.03 | out |
Note: atype of blocker molecule; btotal energy, kcal/mol, of ligand binding by the E protein;
cenergy of van der Waals interactions, dcolumbic interactions, eH-bond energy, fH-bond donor, gH-bond acceptor; idrug binding position, in or out of E channel, in channel blocking position is shown in bold.
Table 1: Binding positions ordered by binding energy of DABCO derivatives with E protein of SARS-cov-2 virus.
Polycyclic derivatives
Polycyclic derivatives of DABCO consists of a large number of atoms, compare to DABCO itself, and will have large van der Waals interactions with inside surface of E channel, PDB code 5X29. A set of molecular blocker have been considered here, molecular structure of which includes large number of atom, than DABCO, and their structure are based of the structures of molecules diazabicyclooctan-benzene (DABCOB), (Figure 3b), and diazabicyclooctan-naphthalene (DABCON), (Figures 3c and 3d). Also, we suggest here two new molecules with larger number of atoms and 3D structure with equal size in three directions. The new molecules are diazabicyclooctan-3-benzene (DABCO3B) and diazabicyclooctan-3-toluene (DABCO3N), (Figures 3c and 3e). Molecules DABCO3B and DABCO3N are obtained from root molecule DABCO by its symmetrical modification in all three directions of the root molecule DABCO, (Figures 4a to 4e). It should be noticed, that construction of inhibitor of E channel using a rigid polycyclic skeleton is preferable compare to the use of a flexible frame, owing to a smaller loss of conformational entropy due to a restriction of conformational mobility of the drug molecule when it is bound inside the E channel. The results of docking of bicyclic and tricyclic derivatives of DABCO onto the E protein are shown in (Figures 4a to 4e and Table 1). Note that (Figures 4a to 4e) show the channel, i.e. residues 8-38 of each of five strands A, D, C, D, E of the E protein, PDB code 5X29. While, docking modeling considered the full structure of E protein as it is defined by the PDB file 5X29.

Figure 3(b): Structure of drug molecule DABCOB.

Figure 3(c): Structure of drug molecule DABCO3B.

Figure 3(d): Structure of drug molecule DABCON.

Figure 3(e): Structure of drug molecule DABCO3N.

Figure 4(a): Structure of complex with drug molecules bound to internal surface of the E channels DABCO.

Figure 4(b): Structure of complex with drug molecules bound to internal surface of the E channels DABCOB.

Figure 4(c): Structure of complex with drug molecule bound to internal surface of the E channels DABCO3B.

Figure 4(d): Structure of complexes with drug molecules bound to internal surface of the E channels DABCON.

Figure 4(e): Structure of complex with drug molecules bound to internal surface of the E channels DABCO3N.
Docking results of DABCOB is shown in (Figure 4b) and results for molecule DABCO3B, is shown in (Figure 4c). The Table 1 indicate that there are two binding modes, namely, in-channel, i.e. drug molecule bound inside the E channel and out of channel, when drug molecule bound to outside molecular surface of the E channel. (Table 1 and Figure 4) show that the binding energies of DABCOB are larger for out of channel mode, by ~5 kcal/mole. The large out of channel binding energy EGAP=EOUT × EIN ~5 kcal/mole, guarantee that the ratio of fractions CIN and COUT of drug molecules bound inside and outside of the E channel bound molecules is negligible (CIN/COUT)<10-2. Thereby the DABCOB drug molecule has extremely low does ability to block E channel.
Docking results of DABCO3B molecule shows, (Table 1), that DABCO3B can be bound in channel and out of channel modes with E channel molecular structure, PDB code 5X29, with a high and practically equivalent binding energies, -30.9 and -30.7 kcal/ mole, and practically equal probabilities ~0.5, (Table 1). The productive in channel and unproductive out of channel binding modes have large binding energies that assumes a productive inchannel binding at low concentrations of drug molecules. (Figure 4c) shown in channel bound molecule with two strong H-bonds, (Table 1). There by, a symmetrical increase of drug molecule sizes in the all three directions of the DABCO, (Figure 3a), increase considerable an affinity of in channel binding and binding energy by more than 5 kcal/mole. The detailed energies of different type of interactions of DABCO3B molecule with E channel are shown in (Table 1). It can be noticed, a large increase, ~14 kcal/mole, of van der Waals interactions of drug molecule with inside surface of E channel.
Results of docking of the largest drug molecule DABCON to the E are shown in (Table 1 and Figure 4d). The binding energy inchannel is large by 1.3 kcal/mole then out of channel modes, i.e. the ratio of the respective fractions of drug molecules CIN/COUT ~0.1. The best in-channel binding affinity has drug molecule DABCO3N, of the largest size, (Figure 3e). The energy of binding of DABCO3N molecule for in-channel mode is the largest one compare to the all other drug molecules considered here, ~-34.7 kcal/mole. The maximal binding energy for out of channel mode is less by ~4 kcal/mole, ~-30.9 kcal/mole, i.e. fraction of out of channel bound drug molecules is negligible, less than 1%. The in channel bound made is characterized by two strong H-bonds with energy ~-9.1 kcal/mole and large van der Waals energy of interactions between drug and E channel interactions. Energy of electrostatic interactions are also favorable, ~-6.8 kcal/mole, compare to the all other types of drug molecules (Table 1).
This paper describes results of construction a drug molecule effective for blocking E channel of virus SARS-cov-2 to destroy and disrupt the functional cycle of virus. An experimental NMR structures, consists of 16 structures which are reflecting thermal dynamics of E channel at physiological conditions. A set of drugs, derivatives of DABCO, were constructed, and their binding affinity to the E protein channel were investigated. The most effective molecular blocker DABCO3N of E channel was determined. The most interesting properties of the new blocker molecule are its abilities to bind tightly via van der Waals interactions and forming two strong H-bonds with two residues, Asn15C-OD1∙∙∙HN1 and Glu8E-OE1H∙∙∙HN2 providing large, more than 34 kcal/mole, binding energy.
This research was supported by Russian Science Foundation grant #21-14-00018 and Russian-Government–funded budget project for the Institute of Chemical Biology and Fundamental Medicine of the Siberian Branch of the Russian Academy of Sciences: #АААА-А17-117020210022-4.
The authors declare that they have no competing interest.
The authors would like to extend their sincere gratitude to Institute of Chemical Biology and Fundamental Medicine of SB RAN.
Citation: Vorobjev NY (2021 ) Design of Effective Molecular Blocker of E Protein Channel as Anti SARS-Cov-2 Virus Drug. Drug Des. 10:186.
Received: 02-Jun-2021 Accepted: 16-Jun-2021 Published: 23-Jun-2021 , DOI: 10.35248/2169-0138.21.10.e186
Copyright: © 2021 Vorobjev NY. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.