Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Review Article - (2014) Volume 3, Issue 2
Polyaromatic hydrocarbons, heterocyclic aromatic amines and dioxin-like compounds are environmental carcinogens shown to initiate cancer in a number of tissue types including prostate and breast. These environmental carcinogens elicit their effects through interacting with the aryl hydrocarbon receptor (AhR), a ligand activated transcription factor. Naturally occurring compounds found in fruits and vegetables shown to have anti-carcinogenic effects also interact with the AhR. This review explores dietary and environmental exposure to chemical carcinogens and beneficial natural compounds whose effects are elicited by the AhR.
<Keywords: Aryl hydrocarbon receptor; Polyaromatic hydrocarbon; Dioxin-like compound; Heterocyclic aromatic amine; Flavonoid; Stilbene; Curcumin
Environmental and dietary exposures are considered risk factors for prostate and breast cancer. Excluding basal cell and squamous cell skin cancers; prostate and breast cancers are the 2 most commonly diagnosed cancer types and account for 28% of all new cancer cases [1]. Compared to cancer incidence in 2003, there is a slight increase in the number of new prostate and breast cancer cases without any significant change in death rates [2]. The increase of new cancer cases may be attributed to exposure to chemical carcinogens.
The relationship between diet and cancer incidence has been a major topic of cancer prevention. Studies showing an association between meat intake and prostate cancers have been inconclusive. Some studies reveal red meat is positively associated with increased prostate cancer risk with an association with more aggressive disease states [3-5]. Despite some studies showing a 43% elevation in prostate cancer risk with high consumption of red meat, others show no association with prostate cancer risk [6-8]. There are also conflicting reports concerning the association of red meat and breast cancer. Both case control and cohort studies have revealed an increased risk of breast cancer associated with meat consumption [9,10]. Conversely, others show no association [11]. Although the role of red meat in prostate and breast cancer remain inconclusive, one explanation for the possible associations reported is the accumulation of carcinogens during the cooking process.
Joshi et al. [12] showed that high fish intake was associated with an increased risk of advanced prostate cancer only when cooked at high temperatures. There was no increased risk for men who consumed fish cooked at low temperatures. Similar associations were found with red meat and poultry cooked at high temperatures. While poultry cooked at low temperatures showed an association with decreased prostate cancer risk [13]. Furthermore, data obtained via food frequency questionnaires revealed that consumption of French fries, fried chicken and fried fish at least once a week was associated with an increased risk of prostate cancer [14]. Consumption of well-done red meat was also associated with a significantly elevated risk of breast cancer within a population based case control study [15,16]. Although, some epidemiologic reports indicate no association with cancer risk and foods cooked at high temperatures, a vast majority have shown that high intake of welldone meat prepared at high temperatures may increase the risk of human cancers [17,18]. Despite inconsistent results of epidemiological studies addressing a link between diet and cancer incidence, the production of carcinogens in certain food types is well established. Additionally, the ability of these food carcinogens to induce prostate and breast cancer has been widely studied.
It has long been thought that diet has an influence on cancer development, and part of the risk may be associated with the consumption of mutagenic substances along with the foods. Several compounds, either present as dietary components or formed during food processing, can play a role in cancer risk [19]. After ingestion most food mutagens go through metabolic activation or detoxification by different endogenous enzymes. Most mutagens begin their adverse effects at the DNA level by forming DNA adducts with carcinogenic metabolites.
Polyaromatic Hydrocarbons (PAHs), heterocyclic aromatic amines and dioxin-like compounds are human carcinogens produced in the meat cooking process. Cooking experiments have shown that certain dioxinlike compounds are produced during cooking at high temperatures [20]. Biological monitoring revealed consumption of grilled, roasted or boiled meat significantly elevated levels of PAHs [21]. Continuous and high temperature grilling was shown to directly contribute to both increased PAHs and heterocyclic aromatic amines accumulation in both fish and beef [22]. Inhalation in certain occupational settings is also a source of human exposure to environmental carcinogens. PAHs are environmental toxicants that are derived from incomplete combustion of organic material such as coal, wood, gasoline and tobacco. They are released into the environment during industrial processes such as paper manufacturing and waste incineration. Dioxin-like compounds are produced as byproducts of incomplete combustion of organic substances in contact with chlorine. They are produced during waste incineration, pulp manufacturing and other industrial processes [23]. Aromatic amines are formed from amino acids, creati (ni) ne and sugar during cooking of fish and meat at high temperatures. The amount of aromatic amines formed varies depending on the cooking time, meat type and cooking method as well as temperature. Certain aromatic amines are also formed during combustion of organic material and are present in diesel exhaust particles and tobacco smoke [24,25]. Dietary consumption is the major sources of exposure in the general population to these environmental carcinogens [26]. PAHs and aromatic amines undergo activation by phase I and phase II drug metabolizing enzymes to be carcinogenic [27]. Compounds within these chemical classes interact with the aryl hydrocarbon receptor (AhR).
AhR is historically known for its role in mediating the toxic and carcinogenic effects of a wide range of environmental contaminants [28,29]. It is a ligand-activated transcription factor that belongs to the basic helix-loop-helix (bHLH), Per-ARNT-Sim (PAS) superfamily of transcription factors [30]. The AhR protein is predominantly cytoplasmic in the majority of normal tissues and binding to exogenous ligands such as PAHs, aromatic amines and dioxin-like compounds, leads to conformational changes that result in nuclear translocation of AhR and dimerization with the AhR nuclear translocator protein (ARNT) [31,32]. The heterodimer binds to a consensus DNA sequence xenobiotic responsive element (XRE) on the enhancer regions of target genes and increases their transcription. These target genes include the cytochrome P450-1 (CYP1) family of genes, which encode enzymes responsible for activation of chemical carcinogens [33,34]. Activation of AhR leads to induction of CYP1A1, CYP1A2 and CYP1B1 genes, which encode for enzymes that metabolize PAHs to mutagenic intermediates resulting in cancer initiation [34-36]. Ligand-dependent activation of AhR not only plays a role in tumor initiation but also in tumor progression [37-39]. Following transcriptional activation, the AhR is exported back to the cytoplasm where it is degraded by calpains and proteasomes [40,41]. Substantial evidence has shown that PAH-dependent activation of AhR plays a role in a variety of cancers including those in breast, liver and lung [35,42].
There is evidence from several labs suggesting that AhR may function as a tumor suppressor gene that becomes silenced during the process of tumor formation under certain conditions [43]. AhR was significantly repressed in tumors from both mice with a liver-specific retinoblastoma protein ablation and their wild-type littermates, supporting the concept that AhR silencing may be associated with cancer progression [44]. Activation of AhR by ligands can inhibit multiple aspects of the metastatic process in a panel of breast cancer cell lines. Dioxin induced protection against breast cancer may occur via down regulation of CXCR4 and CXCL12, thereby inhibiting progression of the disease, regardless of estrogen receptor, progesterone receptor, or human epidermal growth factor receptor 2 status [45]. AhR and its agonists may confer protective effects in multiple breast cancer subtypes by inhibiting invasive and metastatic features and inducing differentiation. These results support previous epidemiological and rodent studies showing a decrease in breast cancer incidence after exposure to AhR agonists. Another study reported that N-nitrosobutyl (4-hydroxybutyl) amine (BBN) can induce bladder cancer via suppression of AhR signaling pathway [46]. Fritz et al. [47,48] found that AhR-null (AhR−/−) (60%), (AhR+/−) (43%) transgenic adenocarcinoma of the mouse prostate (TRAMP) mice developed prostate tumors with greater frequency than AhR wild-type (AhR+/+) (16%) TRAMP mice. This suggests that AhR inhibits prostate carcinogenesis. Hirabayashi and Inoue [49] also found that wild- type (AhR+/+) mice showed a significant extension of lifespan in a gene-dosage-dependent manner with a delayed onset of leukemogenicity than AHR-deficiencies (AhR+/−, AhR−/−) mice. Those data implied that AhR acts as a tumor suppressor gene under some conditions, but the underlying molecular mechanism is still unknown. Future studies are necessary to investigate the potential cell/molecular mechanisms through which the AhR regulates carcinogenesis, and how the AhR contribute to progression or prevention different kinds of tumors. Given the current challenges in treating aggressive metastatic breast cancer, the clinical development of selective AhR modulators may provide an effective, broad-based alternative to current adjuvant therapies [50].
AhR has been shown to influence a number of cellular processes including differentiation, proliferation and cell cycle progression [51-56]. Activation of the receptor by chemical carcinogens has been reported to antagonize androgen receptor signaling. For example AhR ligand, 2,3,7,8-tetrachlorodibenzo-para-dioxin (TCDD) inhibited testosterone-dependent transcriptional activity and testosteroneregulated prostate specific antigen (PSA) expression in a dose dependent manner [57]. TCDD was also shown to block androgen dependent proliferation of prostate cancer cells [58]. Morrow et al. [59] demonstrated that the simultaneous activation of AhR and androgen receptor with TCDD and an androgen derivative respectively, decreased androgen receptor protein levels. This observation has been contributed to the ability of AhR to promote the proteolysis of androgen receptor through assembly of an ubiquitin ligase complex in which AhR acts as a substrate-recognition subunit to recruit the androgen receptor. This action may also explain the antiandrogenic actions of a number of PAHs.
Studies concerned with intrinsic functions of AhR have found that the receptor may promote carcinogenesis. AhR protein and mRNA expression is associated with phases of rapid proliferation and differentiation in certain tissues. AhR-defective cell lines demonstrate a reduced proliferation rate [60]. Ectopic over expression of AhR in immortalized normal mammary epithelial cells induced a malignant phenotype with increased growth and acquired invasive capabilities [61]. A separate study using a constitutively active AhR construct lacking a ligand binding domain revealed that AhR acts as a transcriptional coregulator for the unliganded estrogen receptor. These studies showed that the endogenous estrogen receptor along with the constitutively active AhR was recruited to estrogen- responsive elements to initiate signaling in an androgen depleted environment [62].
Several independent studies have confirmed elevated levels of AhR expression in malignant mammary tissue [63,64]. Elevated levels of AhR proteins were found in highly malignant breast cell lines compared to the lower basal expression found in immortalized and primary human mammary epithelial cells and breast cancer cell lines derived from early stages [64]. A separate study also demonstrated increased AhR protein expression in an advanced prostate cancer cell line compared to a less aggressive isogenic pair [65]. Together, evidence suggest that in advanced prostate and breast cancer AhR may function in cancer progression by mechanisms other than mediating the carcinogenic effects of a number of environmental toxins including PAHs, aromatic amines and dioxin-like compounds. Apart from these environmental contaminants, the AhR can bind with a variety of structurally diverse chemicals found in plants. Several investigators have observed that different dietary components, e.g., flavonoids, resveratrol, curcumin etc., bind to the AhR and exert antagonistic activity [66,67] (Figure 1).
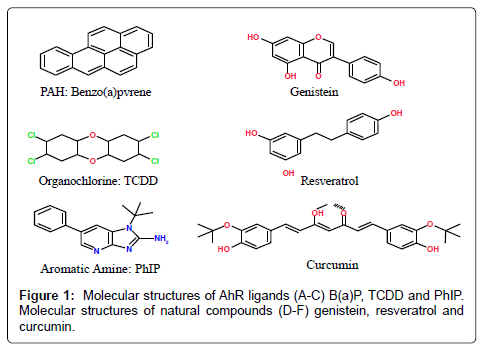
Figure 1: Molecular structures of AhR ligands (A-C) B(a)P, TCDD and PhIP. Molecular structures of natural compounds (D-F) genistein, resveratrol and curcumin.
PAHs are major class of environmental carcinogens and one of major health concern [68]. PAHs refer to a ubiquitous group of several hundred chemically related, environmentally persistent organic compounds of various structures and variable toxicity. PAHs are a class of lipophilic chemicals that consist of fused aromatic rings. They are solely composed of carbon and hydrogen atoms, containing two or more single or fused aromatic rings with a pair of carbon atoms shared between rings [69]. Characterized by their high hydrophobicity, PAHs are also highly resistant to natural degradation. Sources of PAHs can be both natural and anthropogenic and are largely produced as a result of incomplete combustion of hydrocarbon-containing fuels. The main anthropogenic sources of PAHs include open fires, engine exhaust emissions, manufactured gas plants by-products and domestic heating systems, and other organic substances such as tobacco and different food items [70,71]. Human exposure to these compounds can occur by the ingestion of foods, that have been contaminated by water, air, soil, industrial processing or cooked by different methods (frying, smoking, curing). The long-term intake of PAHs represents a health hazard, since they are considered potentially genotoxic and procarcinogenic [71]. PAH exposure has been linked to numerous cancer types including prostate, breast, skin, bladder and lung cancer [72,73]. The Internal Agency for Research on Cancer has classified PAH mixtures as carcinogens to humans [74,75].
The predominant source of PAHs exposure for the non-smoking general population in developed countries is the daily consumption of PAH-contaminated food. The different PAHs levels in food originate from various food processing technologies and home cooking procedures such as barbecuing meat and fish [76,77]. Grilling or broiling of meat, fish or other foods over a direct flame leads to fat dripping on hot fire and yielding gleams containing PAHs that deposit on the surface of the food materials [19,70].
The existence of PAHs has been confirmed in a wide variety of plants and aquatic organisms. However, leafy vegetables are a trivial source of PAHs in the human diet; the level of contamination is overseen by where the vegetables are grown, those situated close to roads or factories are likely to be contaminated with PAHs [76-79]. Plants may absorb PAHs from the air and this could be a more significant factor than PAHs accumulation through root absorption from soil. Also, wastewater treatment plants play an important role in reducing PAHs concentration in wastewater [80]. Incomplete combustion of hydrocarbon-containing fuels produces PAHs which can contaminate the air and water whereby plants can absorb it. In general, humans readily absorb PAHs into the body through the lung, gastrointestinal tract, and skin [81].
Several studies have confirmed the role of AhR in PAH induced toxicities and carcinogenesis. PAH exposed dams revealed extensive branching and enlargement of vessels accompanied by increased expression of antiapoptotic proteins and decreased expression of proapoptotic proteins. AhR-null fetuses did not exhibit these PAHinduced growth alterations [82]. Furthermore, PAH suppression of testicular function, especially spermatogenesis and sperm motility were absent in AhR deficient mice [83]. Curran et al. [84] confirmed the presences of increased levels of PAHs in AhR-null pups demonstrating the importance of AhR-mediated expression of cytochrome P450s in detoxification. Shimizu et al. [85] determined that AhR is required for PAH tumor induction. The prototypic PAH, Benzo [a] pyrene (BaP), induced expression of CYP1A1 in the skin and liver of AhR positive mice and did not induce CYP1A1 expression in AhR-null mice. All AhR positive mice exposed to BaP developed subcutaneous tumors at the site of injection. However, there were no noticeable tumors in the AhR-null mice. These experiments confirmed the carcinogenic action of BaP is mediated by the AhR. BaP is the best-characterized PAHs compound found in diet [19,86]. BaP is rated as carcinogenic to humans by the International Agency for Research on Cancer [87]. This five-ring PAH is present in virtually all PAH mixtures, and is one of the most carcinogenic of those commonly detected. BaP is a mutagenic environmental pollutant, which is suspected to contribute to several types of human cancers [88]. Its carcinogenicity is attributed primarily to its genotoxicity [89]. Although AhR-null mice are protected from BaP induced carcinogenesis, higher levels of BaP-DNA adducts are formed within them than in wild- type mice. AhR positive mice have an effective clearance of BaP metabolites which results in reduced levels of DNA adducts. The lack of a functional AhR in null mice, results in slower clearance of BaP and higher levels of DNA adducts [90]. BaP exposure can impair mismatch repair, which is important in carcinogenesis. High exposure to BaP inhibits mismatch repair activity by reducing expression of mismatch repair protein mutS homolog 6 (MSH6) [91]. It has been conclusively demonstrated in laboratory animal studies that BaP is a powerful carcinogen, which readily induces tumors in various tissues such as lung and skin at relatively low doses [87,92-95]. BaP requires metabolic activation to elicit its detrimental effects. The major end product of its diol epoxide metabolic activation pathway is r-7,t-8,9,c-10-tetrahydroxy-7,8,9,10-tetrahydrobenzo [a] pyrene (trans, anti-BaPT). Individual differences in exposure to, and metabolic activation of, carcinogenic PAHs may influence cancer risk. Measurement of PAHs metabolites in human urine could provide a direct way to assess individual differences in susceptibility to PAHrelated cancer; For example, smokers have significantly higher levels of trans, anti-BaPT in their urine than do non-smokers. It may be useful as a direct phenotyping approach to assess individual differences in uptake and metabolic activation of carcinogenic PAHs [81].
Another class of environmental carcinogens for human is aromatic amines which can be classified into monocyclic aromatic amines, polycyclic aromatic amines and heterocyclic aromatic amines [2-amino-3,8-dimethylimidazo[4,5-f] quinoxaline (MeIQx); 2- amino-2,4-dimethylimidazo[4,5-f]quinoline (MeIQ); 2-amino-1- methyl-6-phenylimidazo [4, 5-b] pyridine (PhIP)]. Most heterocyclic aromatic amines (HAAs), many polycyclic aromatic amines, and some monocyclic aromatic amines are mutagenic [96].
The production of dyes and other complex chemicals can produce carcinogenic aromatic amines and exposure to them happened during and by their use as antioxidants in rubber-manufacturing processes [87,97]. Some aromatic amines produce throughout the tobacco burning [98,99] and arise in the cooking oils releases [100]. Some other HAAs are also produced when tobacco is burning in high- temperature [101,102]; but, consumption of well-done cooked meats is the main source of exposure to many HAAs [103,104]. We can find HAAs also in pan-fried residues used for gravies [105,106], and arise in vapors of cooking oils [107] as well in the air during the frying or grilling of meats [108]. By using experimental laboratory animals and exposing them long-term by carcinogen bioassays, scientists found chemicals from both classes of compounds can initiate tumors at multiple sites. Certain aromatic amines are classified as human carcinogens (Group 1), and some HAAs have been recorded as probable or possible human carcinogens (Group 2A and 2B) [98,109].
HAAs for producing arylnitrenium ion (major metabolite implicated in toxicity and DNA damage) should undergo metabolic activation by N-hydroxylation of the exocyclic amine group [110,111]. During common household cooking, more than 25 HAAs have been shown to form in meats, fish, and poultry [104,112]. The concentrations of HAAs which can be created have a range from around 1 ppb (parts per billion) to more than 500 ppb [103,104,113-115]. The type of meat and the method of cooking also temperature and the duration of cooking are important factors which have effect on amount of HAAs formation during cooking [114,116]. Two major classes of HAAs which produce during heat processing of muscle foods are “pyrolytic HAAs” and “aminoimidazoarenes (AIAs)”. Pyrolytic may arise during the high-temperature pyrolysis (>250°C) of some individual amino acids, including glutamic acid and tryptophan, or during the pyrolysis of proteins [101,103,117], similarly at the low ppb concentrations, pyrolytic HAAs can form, in some cooked meats [118]. On the other hand, AIAs are formed in meats that are cooked at lower temperatures (150-250°C) more usually used in household kitchens. One of the most important way for formation many AIAs is the Maillard reaction (form of nonenzymatic browning resulting from a chemical reaction between an amino acid and a reducing sugar, usually requiring heat) [104,119,120]. During Maillard reaction N-methyl-imidazole-2-ylamine (portion of the molecule) can produce from creatine, and the other portions of the AIAs are supposed to arise from pyridines or pyrazines degradation [119,121]. An aldol condensation is thought to link the two molecules, to form 2-amino-3-methylimidazo [4,5-f] quinoline (IQ) and 2-amino-3- methylimidazo [4,5-f]quinoxaline (IQx)-ring-structured HAAs [122]. It should be noted that the presence of carcinogens in human’s food generation during frying and grilling, has been showed in the number of epidemiological studies. Exposure to meat carcinogens like HAAs or PAHs may increase the risk of a number of common cancers such as breast, prostate and colorectal cancer [4,18,123]. 2-amino-1- methyl-6-phenylimidazo [4,5-b]pyridine (PhIP) is one of the most abundant HAAs detected in cooked meat.
PhIP can form in a model system containing phenylalanine, creatinine, and glucose [124]. However, PhIP can also form in the absence of sugar [104,122]. PhIP is formed in well-done cooked meats and poultry, where the concentration can reach up to 500 ppb [104,114-116,118,120,125].
The most significant gene expression changes in response to PhIP and MeIQx concern members of the AhR gene battery, including CYP1A1 and CYP1A2, which encode two enzymes closely involved in HAAs bio activation [126]. In addition, a number of genes with lower fold changes, including cancer- related genes, whose expression was differentially targeted by PhIP and MeIQx, were observed. HAAs may act in concert with other AhR-activating chemicals found in significant amounts in food and the environment, including PAHs [103,126].
Meat consumption, particularly red and processed meat consumption, has been linked to the increased risk of colorectal cancer in many epidemiological studies [127]. Mutagens such as HAAs and PAHs are formed during high-temperature cooking of meats [128]. These compounds are mutagenic in Ames/Salmonella assays and cause colon tumors in laboratory animals [129]. To exert their mutagenic action, HAAs require enzyme-catalyzed activation consisting of N-oxidation by hepatic CYP1A2 and other extrahepatic P450 isozymes, followed by O-acetylation by N-acetyltransferase 1 (NAT1) and 2(NAT2) [130]. The AhR is an important mediator for xenobiotic signaling to enhance the expression of phase I and II enzymes which affects HAAs metabolism [131].
In order to better understand the molecular basis of HAAs toxicity, Dumont et al have analyzed gene expression profiles in the metabolically competent human Hepa RG cells using pangenomic oligonucleotide microarrays, after either a single (24 hr) or a repeated (28-day) exposure to 10 μM PhIP or MeIQx. The most responsive genes to both HAAs were downstream targets of the AhR: CYP1A1 and CYP1A2 after both time points and CYP1B1 and ALDH3A1 after 28 days. Accordingly, CYP1A1/1A2 induction in HAAs-treated Hepa RG cells was prevented by chemical inhibition or small interference RNA-mediated down-regulation of the AhR. Consistently, HAAs induced activity of the CYP1A1 promoter, which contains a consensus AhR-related xenobiotic-responsive element (XRE). In addition, several other genes exhibited both time-dependent and compound-specific expression changes. These changes concerned genes mainly related to cell growth and proliferation, apoptosis, and cancer. These results identify the AhR gene battery as the preferential target of PhIP and MeIQx in Hepa RG cells and further support the hypothesis that intake of HAAs in diet might increase human cancer risk [103,126]. CYP1A2 is one of the major enzymes that bioactivate a number of procarcinogens including heterocyclic aromatic amines/amides and some natural compounds such as aristolochic acids present in several Chinese herbal medicines. Similar to CYP1A1 and 1B1, CYP1A2 is primarily regulated by the AhR [132].
Derivatives of PhIP were shown to be potential carcinogens for the prostate [133]. PhIP induced prostate carcinogenesis in mice carrying humanized CYP1A2, which activates PhIP to a carcinogen. Low grade prostatic intraepithelial neoplasia (PIN) was seen 20 weeks following administration of PhIP and high-grade PIN 30 to 50 weeks after initial dosage. The lesions induced by PhIP administration were androgen receptor positive and featured the loss of expression of the basal cell marker p63 and the tumor suppressor PTEN [134]. These studies show direct induction of prostate carcinogenesis by PhIP.
Dioxin-like compounds are a diverse group of synthetic chemicals such as dichlorodiphenyltrichloroethane (DDT), dieldrin, hexachlorocyclohexane isomers (HCH), toxaphene, polychlorinated biphenyls (PCBs) and dioxin. Dioxins are a class of polyhalogenated compounds and belong to a group of halogenated aromatic hydrocarbons (HAHs), which have a similar chemical structure and biological effects. They include polychlorinated dibenzodioxins (PCDD), polychlorinated dibenzfurans (PCDF) and polychlorinated biphenyls (PCB). TCDD is identified as the most potent dioxin and is classified as a class I human carcinogen that has been implicated in a number of cancers [135,136]. TCDD exposure was shown to produce quinonoid metabolites of estrogen and the subsequent formation of oxidative DNA lesions through alteration of CYP1A1 and CYP1B1 expression in human breast cancer cells [137]. Kang et al. [138] demonstrated that TCDD significantly increased BaP- DNA adduct formation in the absence of BRCA1. These results imply that oxidative stress is correlated with increased DNA damage in BRCA1 defective cells. Evidence suggests that TCDD might increase the risk of tumorigenesis in BRCA1 defective breast epithelial cells.
Dioxins are derived from the combustion process (e.g., incineration and burning of fuels), during production and utilization of chlorinated compounds (e.g., PCBs) and bleaching of paper-pulp. PCBs enter the air, water and soil during manufacturing, use and disposal. The common features of all these above-mentioned compounds are their persistence in the environment, their bioaccumulation in adipose tissue and in food chains due to their lipophilic character, and their resistance to metabolism. Humans are exposed to dioxins mainly through the consumption of contaminated foods [139]. Among these compounds, TCDD is the most persistent lipophilic environmental contaminant (half-life ~7years) and considered as the prototype chemical [19].
TCDD has the ability to bind to and activate the ligand-activated transcription factor, AhR. Structurally related compounds that bind to the AhR and exhibit biological actions similar to TCDD are commonly referred to as dioxin-like compounds. Ambient human exposure to dioxins occurs through the ingestion of foods containing residues that bioconcentrate through the food chain. The main sources of TCDD released into the environment are from metal smelting, refining, and processing; combustion and incineration sources; chemical manufacturing and processing; biological and photochemical processes; and existing reservoir sources that reflect past releases [140]. Toxic chemicals like TCDD, BaP and PCBs can activate AhR, which subsequently induce CYP1A1 and CYP1B1 expression. Interestingly, estradiol is metabolized by CYP1A1 and CYP1B1, which also activate BaP to reactive DNA-binding intermediates [141-143].
Several classes of beneficial dietary compounds have been described as health-promoting or disease-preventing. Interestingly, many dietary compounds that have chemopreventive properties have been found to also act as antagonist of the AhR signaling pathway. The chemopreventive effect may be due to inhibiting the effects of environmental and food carcinogens. Therefore, dietary ligands could be effective tools in reducing cancer incidence.
Flavonoids are naturally occurring polyphenols present in many fruits and vegetables [144]. These polyphenolic compounds have attracted renewed attention as potential anticarcinogens, and the molecular mechanisms of their anticarcinogenic effects and their bioavailability have been extensively explored. The major dietary flavonoids are flavones, flavonols, and flavan-3-ols (catechins), and they play important roles in cancer prevention. After absorption with or without metabolic conjugation, flavonoids are transported to target organs where they exert their anticarcinogenic activity. The molecular mechanisms of the anticarcinogenic effects of flavonoids include their antagonistic effect on the AhR, and regulation of phase I and II drug metabolizing enzymes and phase III transporters.
Experimental evidence suggests that flavonoids modulate signal transduction pathways at each stage of carcinogenesis [145]. Dietary flavonoid 5,7, dimethoxyflavone significantly inhibited BaP-induced adduct formation and CYP1B1 expression [146]. Heiden et al. [147] investigated the genetic-, time-, dose-, species- and tissue-dependent AhR-mediated agonistic/antagonistic activities of three food flavonoids: quercetin, chrysin and genistein. Human hepatoma (HepG2-Luc) and human breast tumour (T-47D- Luc) cells were compared for tissuedependent effects. Rat hepatoma (H4IIE-ULg) and human hepatoma (HepG2-Luc) cells were compared for species-dependent activities. Evidence showed that quercetin, chrysin and genistein act in a time-, dose species- and tissue-specific way. For example, genistein displayed agonistic activities when exposed to rat hepatoma cells during 6h but not after 24 h.
Flavonoids displayed agonistic/antagonistic activities in human breast tumour cells, depending on the exposure time, while in human hepatoma cells, only antagonistic activities of flavonoids were measured. Induction of CYP1A1 by flavonoids proceeds by various mechanisms, including the direct stimulation of gene or mRNA stabilization. Some flavonoids induce CYPs through binding to AhR. Generally, substrates for AhR are planar aromatic compounds with few bulky substituent groups. That might partly explain the activity of flavonoids, which have similar planar structures as AhR ligands.
Other flavonoids have been shown to directly inhibit CYP1A1 activity, commonly demonstrated to be a competitive-type of inhibition, and to affect CYP1A1 transcription. The most abundant flavonoids, flavonols quercetin and kaempferol, are both dietary ligands of the AhR, but they exert different effects on CYP1A1 expression. Treatment of MCF-7 cells with quercetin resulted in a concentration and time dependent increase in the amount of CYP1A1 mRNA. Kaempferol, by itself, does not affect CYP1A1 expression, but it can interact with the AhR, and act as an antagonist of TCDD induced CYP1A1 transcription. Despite the structural similarity between quercetin and kaempferol, their differential effects might be due to the absence of an additional hydroxyl group on kaempferol, preventing it from achieving an optimal fit into the binding site on AhR to produce transcriptional activation. The binding of kaempferol may block the binding of AhR ligands, and thus inhibit the activity of other ligands such as TCDD [148,149].
Prenylflavone, icaritin, suppressed estrogen stimulated cell proliferation and gene expression in breast cancer cells. Icaritin exposure destabilizes estrogen receptor protein and restricts estrogen receptor-positive cell growth through direct interaction with AhR [150]. 4’,5,7-trihydroxyisoflavone, also known as genistein, is considered a major soy isoflavone. Genistein directly interacts with AhR to inhibit cytochrome P450 enzyme expression [150]. Via this direct interaction, genistein decreased viability of prostate cancer cells [151]. 3,4,5-trihydroxystilbene (resveratrol) a natural component of grape skin and wine, is also a dietary ligand for AhR [152]. Activation of AhR in breast cancer cells inhibits estrogen dependent transcription of tumor suppressor, BRCA1. The addition of resveratrol prevents AhR-mediated epigenetic silencing of BRCA-1 by promoter hyperphosphorylation [153]. Resveratrol regulation of AhR signaling is estrogen receptor independent. Resveratrol inhibited dioxin induced CYP1B1 expression in estrogen receptor-positive and estrogen receptor-negative breast cancer cells [154].
Diferuloylmethane, also known as curcumin is a dietary yellow pigment of Curcuma longa. The anti-carcinogenic properties of curcumin have been established but the mechanisms of action are not fully understood [155-157]. Evidence suggests that curcumin’s beneficial effects may be mediated through the AhR. Pretreatment of mice with curcumin inhibited BaP induced CYP1A1 expression. Decreased CYP1A1 transcription was attributed to decrease nuclear translocation and DNA binding of AhR [158]. Curcumin was also shown to induce significant inhibition of androgen receptor expression in a hormone sensitive prostate cancer cell line. Curcumin attenuated phosphorylation of Akt but increased phosphorylation of betacatenin. Also beta-catenin target genes, cyclin D1 and c-myc, were also decreased in these studies, suggesting that curcumin’s inhibitory effect is through modulation of the Wnt/beta-catenin signaling pathway, possibly following direct interaction with AhR [159].
Ligands for the AhR have been shown to influence cell proliferation, differentiation and apoptosis. Typically, environmental toxins increase proliferation and differentiation while inhibiting apoptosis. Conversely, dietary antagonists for AhR inhibit proliferation and differentiation and induce apoptosis (Figure 2). However, the mechanism used by AhR to exert these effects is not known and cannot be attributed to its ability to induce drug metabolizing enzymes. The consumption of diet containing carcinogens, including PAHs, dioxin-like compounds and heterocyclic aromatic amines is associated with increased cancer risk. Increasing evidence suggests the consumption of dietary compounds found in fruits and vegetables can decrease cancer incidence. The aryl hydrocarbon receptor is a highly conserved transcription factor whose activity is regulated positively by environmental toxins and negatively by dietary antagonist. However, more studies are needed to confirm the species and tissue specific role of AhR and the dietary compounds that interact with the receptor in cancer initiation and progression.
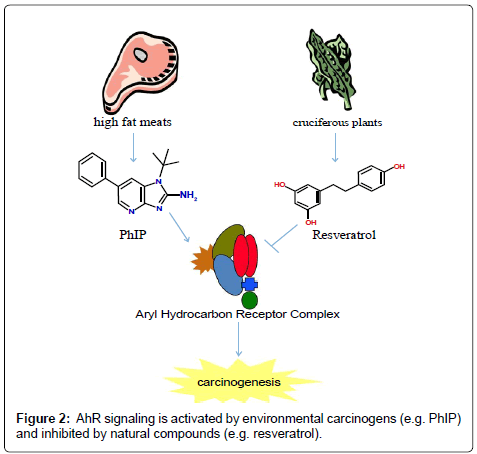
Figure 2: AhR signaling is activated by environmental carcinogens (e.g. PhIP) and inhibited by natural compounds (e.g. resveratrol).